Kepivance
Informace Na Stránce Nepředstavují Lékařskou Radu. Nic Neprodáváme. Přesnost Překladu Není Zaručena. Zřeknutí Se Odpovědnosti
Shrnutí drog
Co je Kepivance?
Kepivance (palifermin) pro injekci je lidský keratinocytový růstový faktor (KGF) produkovaný rekombinantní DNA technologií používanou ke snížení šance na vývoj vředů a vředů v ústech a zkrácení času s vředy nebo vředy u pacientů s rakovinami krve, kteří dostávají vysoké dávky z toho chemoterapie a radiační terapie dříve kostní dřeň transplantace.
Jaké jsou vedlejší účinky kepivance?
Kepivance
- kopřivka
- potíže s dýcháním
- Otok vašeho obličeje rty jazyk nebo krk
- rozmazané vidění
- Vize tunelu
- bolest očí a
- Vidět halos kolem světla
Získejte lékařskou pomoc okamžitě, pokud máte výše uvedené příznaky.
Mezi běžné vedlejší účinky kepivance patří:
- Podráždění kůže (svědění otoku z červeného zaručení)
- pálení nebo pocity brnění v ústech
- Zbarvení jazyka
- zahušťování jazyka
- změny chuti
- otok
- bolest
- bolest kloubů
- Zvýšení enzymů pankreatu břišní
- zvýšený krevní tlak
- protein v moči.
Řekněte svému lékaři, pokud máte nějaké vážné vedlejší účinky kepivance, včetně horečky nebo dýchacích problémů.
Pokud máte následující vážné vedlejší účinky, vyhledejte lékařskou péči nebo zavolejte na číslo 911:
- Vážné příznaky očí, jako je ztráta náhlého vidění rozmazané vidění vidění Vision Vision Eye Eye Eye Eye Eye Eye Eye Eye nebo vidět halos kolem světel;
- Vážné příznaky srdce, jako je rychlé nepravidelné nebo bušení srdečního rytmu; třepování v hrudi; dušnost; a náhlé závratě lightheadness nebo omdlení;
- Těžká zmatek bolesti hlavy zkroucený řečový rameno nebo slabost nohou Potíže se ztrátou chůze pocitu koordinace Pocit nestabilní velmi tuhé svaly vysoké horečky bohaté pocení nebo třes.
Tento dokument neobsahuje všechny možné vedlejší účinky a mohou dojít k jiným. Další informace o vedlejších účincích najdete u svého lékaře.
Dávkování pro kepivance
Doporučená dávka kepivance je 60 mcg/kg/den podávaná jako intravenózní bolusová injekce po dobu 3 po sobě jdoucích dnů před a 3 po sobě jdoucí dny po myelotoxické terapii pro celkem 6 dávek.
Jaké léčivé látky nebo doplňky interagují s kepivací?
Kepivace může interagovat s heparinem. Řekněte svému lékaři všechny léky a doplňky, které používáte.
Kepivace během těhotenství a kojení
Není známo, zda bude kepivance škodlivé pro plod. Řekněte svému lékaři, pokud jste těhotná nebo během léčby otěhotníte. Není známo, zda tento lék prochází do mateřského mléka. Před kojením se poraďte se svým lékařem.
Další informace
Naše kepivance (palifermin) pro injekční vedlejší účinky léky na vedlejší účinky poskytuje komplexní pohled na dostupné informace o léčivě o možných vedlejších účincích při užívání tohoto léku.
Informace o drogách FDA
- Popis léku
- Indikace
- Dávkování
- Vedlejší účinky
- Varování
- Předávkovat
- Klinická farmakologie
- Průvodce léky
Popis kepivance
Kepivance (palifermin) je zkrácený lidský KGF produkovaný technologií rekombinantní DNA v A coli . Kepivace je ve vodě rozpustný 140 aminokyselinový protein s molekulovou hmotností 16,3 kilodaltonů. Liší se od endogenního lidského KGF v tom, že prvních 23 N terminálních aminokyselin byly odstraněny pro zlepšení stability proteinu.
Kepivace je dodávána jako sterilní lyofilizovaný prášek bez bílého konzervačního látek pro intravenózní injekci po rekonstituci s 1,2 ml sterilní vody pro injekční USP. Rekonstituce přináší čirý bezbarvý roztok kepivance (5 mg/ml) s pH 6,5. Každá dávková lahvička kepivance obsahuje palifermin (6,25 mg) s L histidinem (1,94 mg) polysorbátem 20 (0,13 mg nebo 0,01% hm./obj.) A sacharózou (25 mg).
Použití pro kepivance
Indikace
Ukázalo se, že kepivance snižuje výskyt a trvání těžké perorální mukozitidy u pacientů s hematologickou malignitou, které dostávají myelotoxickou terapii při stanovení autologní podpory hematopoetických kmenových buněk. Kepivace je indikována jako podpůrná péče o preparativní režimy, o nichž se předpokládá, že u většiny pacientů bude mít za následek mukozitidu ≥ ≥. Kepivance je mukokutánní epiteliální lidský růstový faktor, který naznačuje ke snížení incidence a trvání těžké perorální mukozitidy u pacientů s hematologickou malignitou, které dostávají myelotoxickou terapii při nastavení autologní hematopoetické podpory kmenových buněk. Kepivace je indikována jako podpůrná péče o preparativní režimy, o nichž se předpokládá, že u většiny pacientů bude mít za následek mukozitidu ≥ ≥.
Omezení použití
Bezpečnost a účinnost kepivance nebyla stanovena u pacientů s nehematologickými malignity). Bezpečnost a účinnost kepivance nebyla stanovena u pacientů s nehematologickými malignity [viz viz Varování a preventivní opatření ].
Kepivance nebyla účinná při snižování výskytu těžké mukozitidy u pacientů s hematologickou malignitou, které dostávaly myelotoxickou terapii při stanovení alogenní podpory hematopoetických kmenových buněk. Kepivace nebyla účinná při snižování výskytu těžké mukozitidy u pacientů s hematologickou malignitou, které dostávaly myelotoxickou terapii při stanovení alogenní podpory hematopoetických kmenových buněk [viz viz Klinické studie ].
Kepivace se nedoporučuje pro použití s melfalanem 200 mg/m jako kondicionační režim. Kepivance se nedoporučuje pro použití s Melphalan 200 mg/m2 jako režim kondicionování [viz Klinické studie ].
Dávkování pro kepivance
Doporučený režim dávkování
Doporučená dávka kepivance je 60 mcg/kg/den podávaná jako intravenózní bolusová injekce po dobu 3 po sobě jdoucích dnů před a 3 po sobě jdoucí dny po myelotoxické terapii pro celkem 6 dávek. Doporučená dávka kepivance je 60 mcg/kg/den podávaná jako intravenózní bolusová injekce po dobu 3 po sobě jdoucích dnů před a 3 po sobě jdoucí dny po myelotoxické terapii pro celkem 6 dávek.
Progesteron pro začátek Vedlejší účinky
Podávejte první 3 dávky před myelotoxickou terapií. Podávejte třetí dávku 24 až 48 hodin před zahájením myelotoxické terapie podávejte první 3 dávky před myelotoxickou terapií. Podávejte třetí dávku 24 až 48 hodin před zahájením myelotoxické terapie [viz Lékové interakce ].
Správa posledních 3 dávek po dokončení myelotoxické terapie; Spravujte první z těchto dávek v den infuze hematopoetických kmenových buněk po dokončení infuze a nejméně 7 dní po posledním podání kepivance [viz viz Lékové interakce ].
Příprava a správa
Příprava
Připravte řešení pro infuzi pomocí aseptické techniky takto:
- Reconstitute kepivance lyofilizovaný prášek sterilní vodou pro injekční USP (není dodáván) pomalu injekcí 1,2 ml sterilní vody pro injekční USP, aby se dosáhla konečné koncentrace 5 mg/ml.
- Během rozpuštění jemně víří obsah. Netřásněte ani energicky agitujte lahvičku. Rozpuštění kepivance může trvat až 3 minuty.
- Před podáním vizuálně zkontrolujte roztok pro zbarvení a částice. Rekonstituovaný roztok by měl být jasný a bezbarvý. Pokud jsou pozorovány zbarvení nebo částice, nespravujte kepivance. Během přípravy nebo podávání nefiltrujte rekonstituovaný roztok. Nezmrzněte rekonstituovaný roztok. Chránit před světlem.
Správa
- Podávejte kepivance intravenózní bolusovou injekcí. Pokud se heparin používá k udržení intravenózní linie, opláchněte linii se solným roztokem před a po podání kepivance [viz Lékové interakce ].
- Rekonstituované řešení neobsahuje žádné konzervační látky a je určeno pouze pro jedno použití. Zlikvidovat jakoukoli nepoužitou část.
- Po rekonstituci se doporučuje, aby byl produkt okamžitě použit. Pokud se nepoužívá okamžitě, může být rekonstituovaný roztok kepivance uložen v jeho kartonu při 2 ° až 8 ° C (36 ° až 46 ° F) po dobu až 24 hodin.
- Před injekcí nechte kepivance dosáhnout teploty místnosti po dobu maximálně 1 hodiny chráněné před světlem. Zlikvidujte kepivace ponechané při pokojové teplotě déle než 1 hodinu.
Jak dodáno
Dávkování Forms And Strengths
Pro injekci : 6,25 mg lyofilizovaného prášku v jednodávkových lahvičkách.
Skladování a manipulace
Kepivance je dodáván jako lyofilizovaný prášek v jednodávkových lahvičkách obsahujících 6,25 mg paliferminu.
Kepivance Vials jsou dodávány v:
- výdejní balíček obsahující 3 lahvičky ( NDC 66658-112-03)
- výdejní balíček obsahující 6 lahviček ( NDC 66658-112-06)
Vložte lahvičky kepivace v dispenční balení v jeho kartonu chlazeném při 2 ° až 8 ° C (36 ° až 46 ° F) až do doby použití. Chránit před světlem.
Vyrobeno: Švédský sirotek Biovitrum AB (Publ) SE-112 76 Stockholm Švédsko. Revidováno: duben 2020
Vedlejší účinky pro kepivance
Zkušenosti klinického hodnocení
Protože klinické studie se provádějí za široce proměnlivých podmínek, nežádoucí rychlosti nežádoucí reakce pozorované v klinických studiích léčiva nelze přímo porovnat s mírami v klinických studiích s jiným léčivem a nemusí odrážet míru pozorované v klinické praxi.
Údaje popsané v tabulce 1 a níže uvedená diskuse odrážejí expozici kepivaci u 409 pacientů s hematologickými malignity, kteří byli zapsáni do 3 randomizovaných klinických studií kontrolovaných placebem a farmakokinetickou studií. Pacienti dostávali kepivace buď před nebo před a po režimech myelotoxiku chemoterapie s nebo bez úplného ozáření těla (TBI) následovanou podporou hematopoetických kmenových buněk. Kepivance byla podávána v denních dávkách v rozmezí 5 až 80 mcg/kg/den. Celková dávka kepivance se pohybovala od 15 do 480 mcg/kg se mediánem 360 mcg/kg. Populace měla střední věk 48 let (rozmezí: 41 až 60 let) 62 % bylo mužů a 83 % bílých s 7,4 % černou a 6,2 % hispánských. Non Hodgkinův lymfom (NHL) byl nejčastější malignitou následovanou Hodgkinovou chorobou mnohočetným myelomem a leukémií.
Nejběžnější nežádoucí účinky přisuzované kepivancí byly toxicity kůže (vyrážky erytému Pruritus) ústní toxicita (dysesteziová jazyk zbarvení zhušťující změnu chuti) Bolest Artralgias a dysestezie. Střední čas na nástup kožní toxicity byl 6 dní po prvním ze 3 po sobě jdoucích denních dávek kepivance se střední dobou trvání 5 dnů. U pacientů, kteří dostávali dysestezii kepivance (včetně hypoestezie a parestezie), byla obvykle lokalizována do periorální oblasti, zatímco u pacientů, kteří dostávali dysestezií placebo, se častěji vyskytly na koncích.
Nejběžnější vážnou nepříznivou reakcí připisovanou kepivaci byla vyrážka kůže hlášená u méně než 1% (3/409) léčených pacientů. Skinské vyrážky stupně 3 se vyskytly u 3% pacientů (9/409), kteří dostávali kepivance a 2% (5/241), kteří dostávali placebo.
Tabulka 1. Výskyt nežádoucích účinků, ke kterým dochází s rozdílem mezi skupinou ≥ 5%
| Tělesný systém Nežádoucí reakce | Kepivance (N = 409) % | PLACEBO (N = 241) % |
| Tělo jako celek | ||
| 28 | 21 | |
| 16 | 11 | |
| 39 | 34 | |
| Gastrointestinal | ||
| 17 | 8 | |
| Muskuloskeletální | ||
| 10 | 5 | |
| Kůže a přívěsky | ||
| 62 | 50 | |
| 35 | 24 | |
| 32 | 22 | |
| Speciální smysly | ||
| 16 | 8 | |
| Centrální nervový systém / periferní nervový systém | ||
| 12 | 7 | |
| Metabolický | ||
| 28 | 23 | |
| 11 | 5 | |
| 62 | 54 | |
| 38 | 31 |
Katarakty
V postmarketingové bezpečnostní studii byl výskyt katarakty numericky vyšší u pacientů, kteří dostávali kepivance než v kontrolní populaci. (vidět Klinické studie ).
Infekce
V randomizované dvojitě zaslepené studii po schválení placebem, která je navržena ke stanovení účinnosti kepivance s vysokou dávkovou melfalanským preparativním režimem, byl výskyt infekcí léčebných podniků významně větší u pacientů léčených kepivancí ve srovnání s placebem. Celkem 281 pacientů bylo randomizováno na 3 ramena: kepivace před melfalanem ve dnech -6 -5 -4 a po Melphalanu ve dnech 0 1 a 2 (pre -post) (n = 115); Kepivace před melfalanem ve dnech -6 -5 -4 (pre) (n = 109); nebo placebo (n = 57). Výskyt hlášených infekcí byl před post -post - 50%; před - 47%; a placebo - 25% [viz Klinické studie ].
Zjištění laboratorních testů: Reverzibilní zvýšení v sérové lipázy a amyláze, které nevyžadovaly léčbu, bylo hlášeno u 28% a 62% pacientů, kteří dostávali kepivance, a 23% a 54% pacientů, kteří dostávají placebo. Obecně byla zvýšení píku pozorována v období cytotoxické terapie a vrácena do výchozí hodnoty ve dne infuze hematopoetických kmenových buněk. Amyláza byla hlavně slinná původ.
Imunogenita
Stejně jako u všech terapeutických proteinů existuje potenciál pro imunogenitu. Klinický význam protilátek vůči kepivaci není znám, ale může zahrnovat sníženou aktivitu a/nebo křížovou reaktivitu s ostatními členy rodiny FGF růstových faktorů.
V klinických studiích byly vzorky séra od pacientů léčených kepivací testovány na protilátky proti kepivaci pomocí elektrochemiluminiscenčního vazebného testu. Dvanáct z 645 pacientů (2%) testovalo pozitivní; Žádná neměla důkaz neutralizační aktivity v buněčném testu.
Výskyt pozitivity protilátky je vysoce závislý na specifickém testu a jeho citlivosti.
Pozorovaný výskyt pozitivity protilátky v testu může být navíc ovlivněn několika faktory, včetně načasování manipulace s vzorkem doprovodných léků a základní onemocnění.
Z těchto důvodů může být srovnání výskytu protilátek proti kepivaci s výskytem protilátek vůči jiným produktům zavádějící.
Zážitek z postmarketingu
Během postgrapovačního používání kepivace v nastavení transplantace kmenových buněk byly identifikovány následující nežádoucí účinky. Protože tyto reakce jsou hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo vytvořit kauzální vztah k expozici léčiva.
- Vaginální edém a erytém;
- Syndrom erythrodysheessia Palmar-Plantar (známý také jako syndrom ruční nohy)
Lékové interakce pro kepivace
In vitro a nadarmo Data ukázala, že palifermin interaguje s nefrakcionovanými a také hepariny s nízkou molekulovou hmotností bez znatelného účinku na farmakodynamiku obou léčiv. Pokud se heparin používá k udržení intravenózní linie, opláchněte linii se solným roztokem před a po podání kepivance [viz Klinická farmakologie ].
Nepodporujte kepivance do 24 hodin před infuzí nebo do 24 hodin po podání myelotoxické chemoterapie [viz viz Dávkování a podávání a Klinické studie ]. In a clinical trial administration of Kepivance within 24 hours of chemoterapie resulted in increased severity a duration of oral mucositis.
Varování pro kepivance
Zahrnuto jako součást 'OPATŘENÍ' Sekce
Opatření pro kepivace
Potenciál pro stimulaci růstu nádoru
Bezpečnost a účinnost kepivance nebyla stanovena u pacientů s nehematologickými malignity. Účinky kepivance na stimulaci nehematopoetických nádorů exprimujících KGF u pacientů nejsou známy. Bylo prokázáno, že kepivance zvyšuje růst buněčných linií lidských epiteliálních nádorů in vitro a to increase the rate of tumor cell line growth in a human carcinoma xenograft model [see Klinická farmakologie ].
Neklinická toxikologie
Zhodnocení mutageneze karcinogeneze plodnosti
Palifermin nebyl karcinogenní ve 26týdenní studii u transgenních myší RAS H2 při intravenózních dávkách 0,1 1 nebo 10 mg/kg/dávka.
Palifermin nebyl genotoxický v reverzní mutační bakteriální (AMES) testování cytogenního testu nebo potkana mikronukleusového testu.
V studii plodnosti a včasného embryonálního vývoje byl palifermin podáván intravenózně mužským a ženským potkanům v dávkách 100 300 nebo 1000 μg/kg/den. Samcům bylo dávkováno 28 dní před pářením soužití a ženy byly podávány 14 dní před pářením skrz den těhotenství 7. Snížený počet epididymálních spermií a zvýšené ztráty po implantaci byly pozorovány v dávkách ≥ 300 μg/kg/den (5krát doporučená lidská dávka na základě tělesné hmotnosti). Zvýšená ztráta před implantací a snížený index plodnosti byla pozorována při dávce paliferminu 1000 μg/kg/den.
Nizorální proti lupům Šampon vedlejší účinky
Použití v konkrétních populacích
Těhotenství
Shrnutí rizika
Na základě zjištění ve studiích na zvířatech může kepivance způsobit poškození plodu při podávání těhotným ženám. O používání kepivance nejsou k dispozici žádné údaje u těhotných žen k informování o riziku závažných vrozených vad a potratu nebo nepříznivých matek nebo fetálních výsledků. Ve studiích reprodukce zvířat intravenózně podávání paliferminu těhotným králíkům a potkanům v období organogeneze vyústilo v úmrtnost na embryo a změny růstu [viz viz Data ].
Odhadované riziko pozadí hlavních vrozených vad a potratu pro uvedené populace není známo. Všechna těhotenství mají na pozadí riziko ztráty vrozených vad nebo jiných nepříznivých výsledků. V americké obecné populaci je odhadované riziko na pozadí hlavních vrozených vad a potratu u klinicky uznávaných těhotenství 2-4% a 15–20%.
Data
Údaje o zvířatech
Ve studiích vývoje embryo-fetálního vývoje byl palifermin podáván intravenózně těhotným králíkům a potkanům během organogeneze. Dávky byly 5 60 a 150 μg/kg/den u králíků (gestační dny 6- 18) a 100 300 a 1000 μg/kg/den u potkanů (těhotenství 6 až 17). Zvýšená ztráta po implantaci a snížená tělesná hmotnost plodu došlo spolu s mateřskou toxicitou (klinické příznaky a snížení přírůstku tělesné hmotnosti a spotřebu potravy) v dávkách 150 μg/kg/den u králíků a 1000 μg/kg/den u potkanů. Zvýšené kosterní změny byly zaznamenány u potkanů při 1000 μg/kg/den. Dávky 150 μg/kg/den u králíků a 1000 μg/kg/den u potkanů jsou přibližně 5krát (králíci) a 35krát (potkanů) expozice (AUC) u pacientů, kteří dostávají doporučenou dávku 60 μg/kg/den.
Laktace
Shrnutí rizika
Neexistují žádné údaje o přítomnosti kepivance v lidském mléce Účinek na kojené dítě nebo účinek na produkci mléka. Protože mnoho léků je vylučováno do lidského mléka a vzhledem k potenciálu vážných nežádoucích účinků u kojení ošetřovatelského dítěte by mělo být během léčby a nejméně 2 týdny po poslední dávce přerušeno.
Ženy a muži reprodukčního potenciálu
Neplodnost
Na základě zjištění ze studií na zvířatech může palifermin narušit plodnost u žen a mužů reprodukčního potenciálu [viz viz Neklinická toxikologie ]. The reversibility of the effects on fertility is unknown.
Dětské použití
Informace o dávkování a bezpečnosti kepivance v dětské populaci jsou omezené. Použití kepivance u pediatrických pacientů ve věku 1 až 16 let je však podporováno důkazy z adekvátních a dobře kontrolovaných studií kepivance u dospělých a studie fáze 1, která zahrnovala 27 pediatrických pacientů s akutní leukemií podstupující hematopoietické kmenové buňky. Byly studovány tři věkové skupiny: věk 1 až 2 (n = 9) ve věku 3 až 11 (n = 9) a věk 12 až 16 (n = 9); 56% bylo mužů 26% bylo kavkazských 63% hispánských; 81% všech 19% AML. Pacienti dostávali vysokodávkovou cytotoxickou terapii sestávající z frakcionovaného celkového ozáření těla (TBI) (12 Gy celková dávka) s vysokou dávkou etoposid (1500 mg/m m2) a vysokou dávku cyklofosfamidu (120 mg/kg) následované alogenní podporou hematopoetických kmenových buněk. Intenzita dávky tohoto přípravného režimu je srovnatelná s intenzitou dávky přípravného režimu 1. vidět Klinické studie . Kepivace byla podávána jako denní intravenózní injekce po dobu 3 po sobě jdoucích dnů před zahájením cytotoxické terapie a po dobu 3 po sobě jdoucích dnů po infuzi hematopoetických kmenových buněk. Byly hodnoceny tři úrovně dávky 40 60 a 80 mcg/kg/dávky. Na žádné úrovni dávky nebyla identifikována žádná toxicita omezující dávku. Nežádoucí účinky byly podobné těm, které byly uvedeny ve studiích dospělých. Výskyt nežádoucích účinků souvisejících s paliferminem byl nejvyšší v kohortě 80 μg/kg. Celkový výskyt perorální mukozitidy WHO byl 10/27 (37%).
Farmakokinetika kepivance byla hodnocena ve studii fáze 1. Věk (1 až 16 let) neovlivnil farmakokinetiku paliferminu v rozmezí dávky (40 až 80 mcg/kg). Koncentrace paliferminu klesly během prvních 30 minut po dávkování. K zvýšení koncentrací paliferminu došlo u některých subjektů přibližně 2 až 4 hodiny po dávce, po kterých následovala druhá fáze pomalého poklesu. Podobný trend byl pozorován u dospělých pacientů. Průměrný rozsah poločasu byl 2,6 až 5,6 hodin u pediatrických pacientů po prvních 60 mcg/kg dávce kepivance. Po 3 po sobě jdoucích dávkách kepivance nebyla pozorována žádná akumulace. Expozice paliferminu se s rostoucími dávkami lineárně nezvýšila. První dávka AUC 0-inf (průměrná) kepivance 60 mcg/kg/den u dospělých pacientů (18 až 63 let) byla 38,2 ng*h/ml ve srovnání s 46,1 ng*h/ml (rozsah prostředků: 22,8 až 81,6) u dětských pacientů (1 až 16 let). Průměrná clearance byla 1730 ml/h/kg pro dospělé a 2481 ml/h/kg (rozsah prostředků: 1700 až 3460) u pediatrických pacientů.
Geriatrické použití
Klinické studie kepivance nezahrnovaly dostatečný počet subjektů ve věku 65 let a starších, aby se určily, zda reagovaly odlišně od mladších subjektů [viz viz Klinická farmakologie ].
Informace o předávkování pro kepivance
Žádné informace
Kontraindikace pro kepivance
Žádný
Klinická farmakologie for Kepivance
Mechanismus působení
KGF je endogenní protein v rodině fibroblastového růstového faktoru (FGF), která se váže na receptor KGF. Bylo popsáno, že vazba KGF na jeho receptor má za následek diferenciaci proliferace a migraci epiteliálních buněk. Receptor KGF Jeden ze čtyř receptorů v rodině FGF byl uváděn přítomen na epiteliálních buňkách v mnoha zkoumaných tkáních, včetně jazyka bukální sliznice, žaludeční střevo slinné žlázy plic jaterních ledvin močové žlázy. Bylo hlášeno, že receptor KGF není přítomen na buňkách hematopoetické linie. Endogenní KGF je produkován mezenchymálními buňkami a je upregulován v reakci na poškození epiteliální tkáně.
U myší a potkanů kepivance zvýšila proliferaci epiteliálních buněk (měřeno imunohistochemickým zbarvením Ki67 a absorpcí BrdU) a prokázala zvýšení tkáňové tloušťky bukální sliznice a gastrointestinálního traktu. Kepivance byla studována na myších modelech chemoterapie a záření vyvolaného gastrointestinálního poškození. U takových modelů podávání kepivance před a/nebo po cytotoxickém urážce zlepšilo přežití a snížilo úbytek hmotnosti ve srovnání s kontrolními zvířaty.
Kepivance has been shown to enhance the growth of human epithelial tumor cell lines in vitro při koncentracích ≥ 10 mcg/ml (> 15krát vyšší než průměrné terapeutické koncentrace u lidí). U nahých myších xenoštěpových modelů byly tři po sobě jdoucí denní ošetření kepivance v dávkách 1500 a 4000 mcg/kg (25- a 67krát vyšší než doporučená lidská dávka) týdně po dobu 4 až 6 týdnů byla spojena se zvýšením růstu v závislém na dávce 1 ze 7 kgf receptorexpresivních buněčných linií nádoru.
Farmakodynamika
Proliferace epiteliálních buněk byla hodnocena imunohistochemickým barvením KI67 u zdravých subjektů. V bukálních biopsiích od 3 ze 6 zdravých subjektů bylo pozorováno 3-ohybné nebo větší zvýšení barvení KI67 při 40 mcg/kg/den intravenózně po dobu 3 dnů, když bylo měřeno 24 hodin po třetí dávce. Proliferace epitelových buněk dávkované dávky byla pozorována u zdravých subjektů podávaných jednotlivými intravenózními dávkami 120 až 250 mcg/kg 48 hodin po dávkování.
Farmakokinetika
Farmakokinetika kepivance byla studována u zdravých subjektů a pacientů s hematologickými malignity. Po jednotlivých intravenózních dávkách 20 až 250 mcg/kg u zdravých subjektů a 60 mcg/kg u pacientů s rakovinou kepivance koncentrace klesly o více než 95% během prvních 30 minut po dávce. Mírné zvýšení nebo náhorní plošinu v koncentraci došlo přibližně při přibližně 1 až 4 hodinách následované fází poklesu terminálu. Kepivace vykazovala lineární farmakokinetiku s extravaskulární distribucí. U pacientů s rakovinou ve srovnání se zdravými subjekty po 60 mcg/kg jediné dávky kepivance byla průměrná celková clearance těla (CL) 2 až 4krát vyšší a objemová distribuce v ustáleném stavu (VSS) byla 2krát vyšší.
Eliminační poločas byl podobný mezi zdravými subjekty a pacienty s rakovinou (průměrně 4,5 hodiny s rozsahem 3,3 až 5,7 hodiny). Po 3 po sobě jdoucích denních dávkách 20 a 40 mcg/kg u zdravých jedinců nebo 60 mcg/kg u pacientů s rakovinou nedošlo k žádné akumulaci kepivance. Věk (1 až 16 let) neovlivnil farmakokinetiku paliferminu v rozmezí dávky 40 až 80 mcg/kg [viz Použití v konkrétních populacích ].
Lékové interakce
Společné podávání s heparinem
Potenciální farmakokinetická interakce mezi paliferminem a heparinem byla hodnocena ve studii s jednou dávkou u 27 zdravých subjektů, kteří dostávali palifermin (60 mcg/kg) společně s terapeutickými hladinami nefrakcionovaného heparinu a bez ní. Tato spolupracovníka vedla k pětinásobnému zvýšení paliferminové AUC a 80% snížení průměrného CL. Palifermin nebyl žádný významný účinek na aktivitu heparinu s ohledem na aktivovaný částečný čas tromboplastinu (APTT). Druhá studie byla provedena u 31 hodnotitelných zdravých subjektů, kteří dostávali palifermin (40 mcg/kg/den po dobu 3 dnů) společně s terapeutickými hladinami nefrakcionovaného heparinu a bez ní. V této studii byla souvislost heparinu a paliferminu vyústila v 425% zvýšení paliferminové AUC a 76,5 73,1 a 38,8% snížení objemu distribuce paliferminu a poločasu. Tyto změny v paliferminu PK neměly znatelný účinek na expresi Ki67 v bukálních biopsiích používaných jako marker proliferace epitelových buněk.
Farmakokinetika In Specific Populations
Poškození ledvin
Výsledky studie farmakokinetiky u 24 subjektů s různým stupněm poškození ledvin prokázaly, že poškození ledvin má malý nebo žádný vliv na farmakokinetiku kepivance.
Poškození jater
Farmakokinetický profil pacientů s jaterní nedostatečností nebyl hodnocen.
Starší
V jedné dávkové studii dostali subjekty 180-mcg/kg nebo 90 mcg/kg dávku paliferminu podávané intravenózní bolusovou injekcí. Subjekty starší 65 let (n = 8) měly v průměru přibližně 30% nižší míru CL než ty 65 a mladší (n = 19). Pro geriatrickou populaci se nedoporučuje žádná úprava dávky [viz Použití v konkrétních populacích ].
Klinické studie
Autologní transplantace Preparativní režimy, které zahrnují celkové ozáření těla
The safety and efficacy of Kepivance in decreasing the incidence and duration of severe oral mucositis in patients with hematologic malignancies (NHL Hodgkin's disease acute myeloid leukemia acute lymphoblastic leukemia chronic myeloid leukemia chronic lymphocytic leukemia or multiple myeloma) receiving myelotoxic therapy requiring hematopoietic stem cell Podpora byla stanovena v randomizované placebem kontrolované klinické studii s 212 pacienty (studie 1) a randomizované klinické studie s rozsahem 169 pacientů s náhodnou rozvrhovou rozhraní (studie 2).
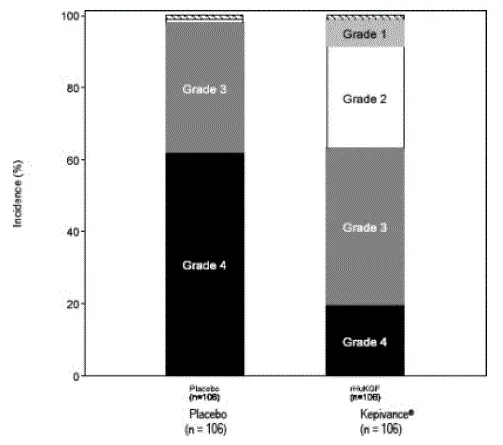
Ve studii 1 pacienti dostávali vysokodávkovou cytotoxickou terapii sestávající z frakcionovaného ozáření celkového těla (TBI) (12 Gy celkové dávky) s vysokou dávkovou etoposidem (60 mg/kg) a vysokou dávkovou cyklofosfamidu (100 mg/kg) následované hematopoetickou kmenovou buňkou. Pacienti byli randomizováni, aby dostávali kepivance (n = 106) nebo placebo (n = 106). Kepivance 60 mcg/kg byla podávána jako denní intravenózní injekce po dobu 3 po sobě jdoucích dnů před zahájením cytotoxické terapie a po dobu 3 po sobě následujících dnů po infuzi hematopoetických kmenových buněk. Hlavním výsledkem účinnosti byl počet dní, během kterých pacienti zažili těžkou perorální mukozitidu (stupeň 3/4 na stupnici WHO [Světové zdravotnické organizace]) 1. Mezi další analýzy patřily doba trvání výskytu a závažnost perorální mukositidy a použití opioidní analgezie. U pacientů, kteří dostávali kepivance ve srovnání s pacienty, kteří dostávali placebo, neexistoval žádný důkaz o zpoždění v čase u pacientů, kteří dostávali kepivaci. Výsledky studie 1 jsou uvedeny v tabulce 2 a obrázku 1.
Tabulka 2: Studie 1 Výsledky účinnosti
| Proměnná účinnost | Kepivance (60 mcg/kg/den) (N = 106) | PLACEBO (N = 106) |
| Medián (25 th 75 th Percentil) Dny WHO stupně 3/4 Orální mukozitida* | 3 (0 6) | 9 (6 13) |
| Výskyt ústní mukozitidy stupně Who 3/4 | 63% (67/106) | 98% (104/106) |
| Medián (25 th 75 th Percentil) Dny o perorální mukozitidě třídy 3/4 u postižených pacientů | 6 (3 8) (n = 67) | 9 (6 13) (n = 104) |
| Výskyt ústní mukozitidy | 20% | 62% |
| Medián (25 th 75 th Percentil) Kumulativní dávka opiodu (morfinové MG ekvivalenty) | 212 (3 558) | 535 (269 1429) |
| * P. <0.001 COMPARED TO PLACEBO USING GENERALIZED COCHRAN-MANTELHAENSZEL (CMH) TEST STRATIFIED FOR STUDY CENTER. |
Obrázek 1: Studie 1 Výskyt perorální mukozitidy o maximální stupeň, který orální mukozitida stupnice
 |
Studie 2 byla randomizovaná placebem ovládaná vícecentka Zkouška porovnávající různé plány kepivance. Všichni pacienti dostávali vysokou dávkovou cytotoxickou terapii sestávající z frakcionované TBI (12cgy celkové dávky) s vysokou dávkovou etoposidem (60 mg/kg) a vysokou dávkovou cyklofosfamid (75-100 mg/kg) následované podporou hematopoetických kmenových buněk. Výsledky pro studii 1 byly podpořeny výsledky pozorovanými u podskupiny pacientů ve studii 2, kteří dostávali stejnou dávku a plán kepivance podávané ve studii 1.
Jedno rameno studie 2, která zahrnovala pacienty, kteří dostávali kepivance po dobu 3 po sobě jdoucích dnů před zahájením cytotoxické terapie, dávka daná poslední den TBI před etoposidem a po dobu 3 po sobě jdoucích dnů po infuzi hematopoietických kmenových buněk byla předčasně uzavřena bezpečnostním výborem pro nedostatek účinnosti a trendem ve srovnání s ústními mucositidy ve srovnání s placemi ve srovnání s placemi ve srovnání s placemi ve srovnání s placemi ve srovnání s placemi v rámci s orálním kmenovým buňkám. Výbor pro bezpečnost přičítal bezpečnostní zjištění kepivaci, která byla podána do 24 hodin od chemoterapie, což mělo za následek zvýšenou citlivost rychle se dělících epiteliálních buněk v bezprostředním období po chemoterapii [viz viz Dávkování a podávání a Lékové interakce ].
WHO ORAL MUCOSITIS SCATER: stupeň 1 = bolest/erytém; Stupeň 2 = vředy erytému mohou jíst pevné látky; Stupeň 3 = vředy vyžadují pouze kapalnou stravu; Stupeň 4 = alimentace není možné.
Nedostatek účinnosti
Autologní transplantace Preparativní režim s použitím vysoké dávky melfalan
Ve studii studie po schvalování 3 byla určena ke stanovení účinnosti kepivance s vysokou dávkovou melfalanským přípravným režimem s více myelomem v multicentrické randomizované dvojitě slepé placebem kontrolované studii. Kondicionační režim byl Melphalan (200 mg/m2) v den -2 následované autologní podporou hematopoetických kmenových buněk. Celkem 281 pacientů bylo randomizováno na 3 ramena: kepivace před melfalanem ve dnech -6 -5 -4 a po Melphalan ve dnech 0 1 a 2 (přípravné) (n = 115); Kepivace před melfalanem ve dnech -6 -5 -4 (pre) (n = 109); nebo placebo (n = 57).
Hlavním výsledkem studie byla maximální závažnost ústní mukozitidy. Střední věk přihlášených pacientů byl 57 let (rozmezí 32-69) a 55% mužů. Výsledky jsou uvedeny na obrázku 2. Prespecifikovaná primární analýza byla srovnáváním mezi předstihem a předem pažemi na placebo.
Výskyt třídy WHO 3 a 4 v rameni před post-post kepivance byl 38% ve srovnání s 37% v rameni s placebem. Nebyly zjištěny žádné významné rozdíly mezi jedním z režimů kepivance a placebo ramenem ve výskytu těžké perorální mukozitidy.
Obrázek 2: Incidence perorální mukozitidy maximální stupeň, který perorální mukozitida ve vysoké dávce
 |
Podskupina subjektů zapsaných do studie mnohočetného myelomu byla zahrnuta do hodnocení rizika vývoje katarakty u pacientů s léčbou kepivance. Oftalmologické vyšetření byly provedeny u 101 pacientů zařazených do dvojitě slepé randomizované placebem kontrolované studie dvou různých rozvrhů kepivance (pouze po chemoterapii a po chemoterapii) pro snížení závažnosti perorální mukozitidy u subjektů s více myelomem přijímajícím translační perifální krevní buňku. Pro koncový bod incidence primárního katarakta vývoje katarakty nebo progresi katarakty ve 12. měsíci došlo k většímu podílu subjektů, které zaznamenaly vývoj katarakty ve skupině Kepivance: 48% (25/52) ve srovnání se skupinou placeba: 29% (4/14) (rozdíl 17 [95% CI: -11 46]) [Viz)) Nežádoucí reakceS ].
Nedostatek účinnosti
Alogenní transplantace
Ve studii po schvalování, která má stanovit účinnost kepivance při snižování výskytu závažného akutního štěpu versus hostitelské onemocnění (AGVHD) u pacientů s hematologickou malignitou podstupující alogenní transplantaci, byla také měřena doba trvání a závažnosti perorální mukositidy. Bylo použito více kondicionačních režimů. Pacienti byli randomizováni do placeba (n = 78) nebo ke kepivaci (n = 77) 60 mikrogramů/kg pro 3 dávky před režimem kondicionování a 180 mikrogramů/kg nejméně 24 hodin od poslední dávky chemoterapie a nejméně 24 hodin před první dávkou po transplantační methotrexatu. Ve srovnání s placebem (17%) nebyl žádný rozdíl ve výskytu závažné agvhd kepivance (16%). Kromě nedostatku účinnosti při prevenci závažné AGVHD byl výskyt mukozitidy WHO 3. a 4 stupně u pacientů léčených kepivancí (81%) ve srovnání s placebem (73%).
Informace o pacientovi pro kepivance
Poraďte pacientům, aby poskytovatelům zdravotní péče nahlásili následující:
- Vyrážky a zčervenání kůže [viz Nežádoucí reakceS ]
- Svědění [viz Nežádoucí reakceS ]
- Otok jazyka [viz Nežádoucí reakceS ]
- Změny v pocitu úst a jazyka [viz Nežádoucí reakceS ]
- Změna chuti [viz Nežádoucí reakceS ]
Informujte pacienty:
- Že bezpečnost a účinnost kepivance nebyla stanovena u pacientů s nehematologickými malignitami [viz Indikace a Varování a preventivní opatření ]
- Důkazů o růstu a stimulaci nádoru v buněčné kultuře a ve zvířecích modelech nehematopoetických lidských nádorů [viz viz Varování a preventivní opatření a Klinická farmakologie ]