Besponsa
Informace Na Stránce Nepředstavují Lékařskou Radu. Nic Neprodáváme. Přesnost Překladu Není Zaručena. Zřeknutí Se Odpovědnosti
Shrnutí drog
Co je to Bespsonsa?
Besponsa (inotuzumab ozogamicin) pro injekci je konjugát protilátek s protilátkou (ADC) zaměřený na CD22 (ADC) pro léčbu dospělých s relapsovaným nebo refrakterním prekurzorem B-buněk akutní lymfoblast leukémie (VŠE).
Jaké jsou vedlejší účinky pro BeSpsonsu?
Besponsa
- kopřivka
- potíže s dýcháním
- Otok vašeho obličeje rty jazyk nebo krk
- Cítím se chladno
- svědění
- horečka
- dušnost
- horečka
- slabost
- Symptomy nachlazení nebo chřipky
- kašel
- oteklé dásně
- vředy
- bledá kůže
- Snadné modřiny
- Vředy
- Bolesti hlavy s bolestí na hrudi a těžkou závratě
- mdloby
- Rychlé nebo bušící srdeční rytmus
- Bolest horního žaludku
- žloutnutí kůže nebo očí ( žloutenka )
- rychlý přírůstek na váze
- otok v pažích nebo nohou
- Bolestivé otoky ve vaší střední části
- krvácející dásně
- Abnormální vaginální krvácení
- krev ve vaší moči nebo stolici
- kašeling up blood a
- zvracení, které vypadá jako káva
Získejte lékařskou pomoc okamžitě, pokud máte výše uvedené příznaky.
Mezi běžné vedlejší účinky BesPsonsa patří:
- Nízký počet destiček (trombocytopenie)
- nízký počet bílých krvinek se počítá ( Neutropenie Leukopenia)
- infekce
- anémie
- únava
- krvácející
- horečka
- nevolnost
- bolest hlavy
- Febrilní neutropenie
- Transaminázy se zvýšily
- Průměr břicha
- zácpa
- zvracení
- otok a boláky uvnitř úst
- zimnice
- Gamma-glutamyltransferáza se zvýšila a
- příliš mnoho bilirubinu v krvi
Pokud máte následující vážné vedlejší účinky, vyhledejte lékařskou péči nebo zavolejte na číslo 911:
- Vážné příznaky očí, jako je ztráta náhlého vidění rozmazané vidění vidění Vision Vision Eye Eye Eye Eye Eye Eye Eye Eye nebo vidět halos kolem světel;
- Vážné příznaky srdce, jako je rychlé nepravidelné nebo bušení srdečního rytmu; třepování v hrudi; dušnost; a náhlé závratě Lightheadedness nebo omdlet;
- Těžká zmatek bolesti hlavy zkroucený řečový rameno nebo slabost nohou Potíže se ztrátou chůze pocitu koordinace Pocit nestabilní velmi tuhé svaly vysoké horečky bohaté pocení nebo třes.
Tento dokument neobsahuje všechny možné vedlejší účinky a mohou dojít k jiným. Další informace o vedlejších účincích najdete u svého lékaře.
Dávkování pro Besponsu
Dávka a délka cyklu pro besponsa závisí na cyklu den cyklu a na jakékoli předchozí reakci na léčbu. Pacienti jsou předem medializováni kortikosteroidní anti-fever léky a antihistaminikum před všemi infuzemi Bespsonsa.
Jaké léky nebo doplňky interagují s BesPsonsou?
Besponsa může interagovat s léky, o nichž je známo, že prodlužují interval QT nebo indukují torsades de pointes. Řekněte svému lékaři všechny léky a doplňky, které používáte.
Besponsa během těhotenství a kojení
Besponsa se nedoporučuje pro použití během těhotenství; Může to poškodit plod. Ženám se doporučuje používat antikoncepci při používání besponsa. Při používání besponsa se kojení nedoporučuje.
Kolik Ativan vás může zabít
Další informace
Naše besponsa (inotuzumab ozogamicin) pro injekční vedlejší účinky drogové centrum poskytuje komplexní pohled na dostupné informace o možných vedlejších účincích při užívání tohoto léku.
Informace o drogách FDA
- Popis léku
- Indikace
- Vedlejší účinky
- Varování
- Předávkovat
- Klinická farmakologie
- Průvodce léky
VAROVÁNÍ
Hepatotoxicita včetně jaterního veno-okluzivního onemocnění (VOD) (také známé jako sinusová obstrukční syndrom a zvýšené riziko transplantace po hematopoetickém kmenovém buňce (HSCT) nerelapse úmrtnosti
Hepatotoxicita včetně VOD
- Hepatotoxicita včetně fatálního a život ohrožujícího VOD se vyskytla u pacientů s relapsovanou nebo refrakterní akutní lymfoblastickou leukémií (vše), kteří dostali besponsu. Riziko VOD bylo větší u pacientů, kteří podstoupili HSCT po léčbě besponsa; Použití režimů kondicionování HSCT obsahujících 2 alkylační činidla a poslední celková hladina bilirubinu ≥ Horní hranice normálního (ULN) před HSCT byla významně spojena se zvýšeným rizikem VOD.
- Mezi další rizikové faktory pro VOD u pacientů léčených besponsou patřily probíhající nebo předchozí onemocnění jater před HSCT zvýšené věk pozdějších linií záchrany a větší počet cyklů léčby besponsa.
- Zvýšení testů jater může vyžadovat snížení dávky dávkování nebo trvalé přerušení besponsa. Pokud dojde k VOD, trvale přerušte léčbu. Pokud dojde k závažnému VOD, léčí podle standardní lékařské praxe [viz Dávkování a podávání a VAROVÁNÍS AND OPATŘENÍ ].
Zvýšené riziko úmrtnosti na relapsu po HSCT
- U pacientů dostávajících BesPsonsa byla vyšší míra úmrtnosti na relapsu po HSCT, což mělo za následek vyšší den po HSCT úmrtnosti 100 [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Popis pro Besponsa
Inotuzumab ozogamicin is a CD22-directed antibody-drug conjugate (ADC) consisting of 3 components: 1) the recombinant humanized immunoglobulin class G subtype 4 (IgG4) kappa antibody inotuzumab specific for human CD22 2) N-acetyl-gamma-calicheamicin that causes double-stranded DNA breaks and 3) an acid-cleavable linker composed of the condensation product of 4-(4’-acetylphenoxy)-butanoic acid (AcBut) and 3-methyl-3-mercaptobutane hydrazide (known as dimethylhydrazide) that covalently attaches N-acetyl-gamma-calicheamicin to inotuzumab.
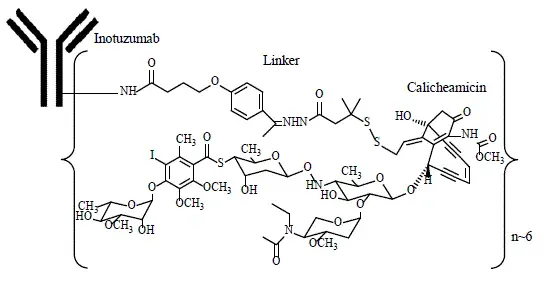 |
Inotuzumab ozogamicin má přibližnou molekulovou hmotnost 160 kDa. Průměrný počet derivátových molekul kalicheamicinu konjugovaných s každou molekulou inotuzumabu je přibližně 6 s distribucí od 2-8. Inotuzumab ozogamicin je produkován chemickou konjugací složek protilátky a malých molekul. Protilátka je produkována buňkami savčích (vaječníků čínského křečka) a semisyntetický derivát kalichemicinu je produkován mikrobiální fermentací a následuje syntetická modifikace.
Besponsa (inotuzumab ozogamicin) pro injekci je dodáván jako sterilní bílá až bělavý lyofilizovaný prášek bez bílého konzervačních látek pro intravenózní podání. Každá dávková lahvička poskytuje 0,9 mg inotuzumab ozogamicin. Neaktivní ingredience jsou polysorbát 80 (0,36 mg) chloridu sodného (2,16 mg) sacharózy (180 mg) a tromhamin (8,64 mg). Po rekonstituci se 4 ml sterilní vody pro injekci USP je konečná koncentrace 0,25 mg/ml inotuzumabu ozogamicinu s výskyt objemu 3,6 ml (0,9 mg) a pH přibližně 8,0.
Použití pro besponsu
Besponsa je indikována pro léčbu relapsovaného nebo refrakterního CD22-pozitivního prekurzoru B-buněk akutní lymfoblastická leukémie (vše) u dospělých a dětských pacientů 1 rok a starší.
Dávkování pro Besponsu
Doporučené dávkování
- Předběžný lék před každou dávkou [vidět Doporučená předškolní a cytoredukce ].
- Spravujte pouze intravenózní infuzí.
- Pro první cyklus je doporučená celková dávka besponsa pro všechny pacienty 1,8 mg/m2 za cyklus podávaný jako 3 rozdělené dávky v den 1 (0,8 mg/m2) Den 8 (0,5 mg/m2) a den 15 (0,5 mg/m2). Cyklus 1 je 3 týdny v délce trvání, ale může být prodloužen na 4 týdny, pokud pacient dosáhne úplné remise (CR) nebo úplné remise s neúplným hematologickým zotavením (CRI) a/nebo aby umožnil zotavení z toxicity.
- Pro následující cykly:
- U pacientů, kteří dosáhnou CR nebo CRI, je doporučená celková dávka BesPonsa 1,5 mg/m2 za cyklus podávaný jako 3 rozdělené dávky v den 1 (0,5 mg/m2) Den 8 (0,5 mg/m2) a den 15 (0,5 mg/m2). Následující cykly jsou po dobu 4 týdnů.
NEBO
- U pacientů, kteří nedosáhnou CR nebo CRI, je doporučená celková dávka besponsa 1,8 mg/m2 za cyklus uvedený jako 3 rozdělené dávky v den 1 (0,8 mg/m2) Den 8 (0,5 mg/m2) a den 15 (0,5 mg/m2). Následující cykly jsou po dobu 4 týdnů. Patients who do not achieve a Cr nebo CRI within 3 cycles should discontinue treatment.
- U pacientů, kteří dosáhnou CR nebo CRI, je doporučená celková dávka BesPonsa 1,5 mg/m2 za cyklus podávaný jako 3 rozdělené dávky v den 1 (0,5 mg/m2) Den 8 (0,5 mg/m2) a den 15 (0,5 mg/m2). Následující cykly jsou po dobu 4 týdnů.
- U pacientů, kteří postupují k transplantaci hematopoetických kmenových buněk (HSCT), je doporučená doba léčby besponsa 2 cykly. Třetí cyklus lze zvážit pro ty pacienty, kteří nedosahují CR nebo CRI a minimálního zbytkového onemocnění (MRD) negativita po 2 cyklech [viz viz VAROVÁNÍS AND OPATŘENÍ ].
- U pacientů, kteří nepostupují k HSCT, může být podáno další cykly léčby až do maximálně 6 cyklů.
Tabulka 1 ukazuje doporučené režimy dávkování.
Tabulka 1. Dávkovací režim pro cyklus 1 a následné cykly v závislosti na reakci na léčbu
| Den 1 | 8. dena | Den 15a | |
| Dávkovací režim pro cyklus 1 | |||
| Všichni pacienti: | |||
| b | 0,8 mg/m2 | 0,5 mg/m2 | 0,5 mg/m2 |
| 21 dníc | |||
| Dávkovací režim pro následující cykly v závislosti na reakci na léčbu | |||
| Pacienti, kteří dosáhli CRd nebo CRIe: | |||
| b | 0,5 mg/m2 | 0,5 mg/m2 | 0,5 mg/m2 |
| 28 dníf | |||
| Pacienti, kteří nedosáhli CRd nebo CRIe: | |||
| b | 0,8 mg/m2 | 0,5 mg/m2 | 0,5 mg/m2 |
| 28 dníf | |||
| Zkratky: Cr = úplná remise; CRI = úplná remise s neúplným hematologickým zotavením. a /-2 dní (udržujte minimálně 6 dní mezi dávkami). b Dávka je založena na povrchu těla pacienta (M2). c U pacientů, kteří dosáhnou CR nebo CRI a/nebo aby umožnili zotavení z toxicity, může být délka cyklu prodloužena až na 28 dní (tj. 7denní interval bez léčby počínaje 21. dnem). d Cr je definován jako <5% blasts in the bone marrow a the absence of peripheral blood leukemic blasts full recovery of peripheral blood counts (platelets ≥ 100 x 109/L a absolutní počty neutrofilů [ANC] ≥ 1 x 109/L) a rozlišení jakéhokoli extramedulárního onemocnění. e CRI je definován jako <5% blasts in the bone marrow a the absence of peripheral blood leukemic blasts incomplete recovery of peripheral blood counts (platelets < 100 x 109/L a/nebo anc <1 x 109/L) a rozlišení jakéhokoli extramedulárního onemocnění. f 7denní interval bez léčby počínaje 21. den. |
Doporučená předškolní a cytoredukce
- Před dávkováním se doporučuje premedikace kortikosteroidní antipyretická a antihistaminikum. Pacienti by měli být pozorováni během a po dobu nejméně 1 hodiny po skončení infuze pro příznaky reakcí souvisejících s infuzí [Viz [Viz VAROVÁNÍS AND OPATŘENÍ ].
- U pacientů s cirkulujícími lymfoblasty cytoredukce s kombinací steroidů hydroxyurea a/nebo vinkristinu na periferní počet výbuchu menší nebo rovné 10000/mm3 se doporučuje před první dávkou.
Modifikace dávkování pro nežádoucí účinky
Modifikujte dávku besponsa pro toxicitu (viz tabulky 2-4). Dávky besponsa v léčebném cyklu (tj. Dny 8 a/nebo 15) nemusí být přerušeny kvůli neutropenii nebo trombocytopenii, ale přerušení dávkování v cyklu se doporučuje pro nehematologickou toxicitu. Pokud je dávka snížena v důsledku toxicity související s BESPSA, dávka nesmí být znovu eskalována.
Tabulka 2. Modifikace dávkování BespOnsa pro hematologické toxicity [viz viz VAROVÁNÍS AND OPATŘENÍ ]
| Kritéria | Modifikace dávkování besponsa |
| Pokud byl před léčbou BeSpsonsa ANC větší nebo roven 1 x 109/L | Pokud se ANC sníží, přerušte další cyklus léčby až do zotavení ANC na větší nebo rovné 1 x 109/L. Discontinue Besponsa if low ANC persists fnebo greater than 28 dní a is suspected to be related to Besponsa. |
| Pokud byl před léčbou besponsa počet krevních destiček větší než 50 x 109/La | Pokud se počet krevních destiček sníží, přerušte další cyklus léčby, dokud se počet destiček nezotaví na větší nebo rovný 50 x 109/La . Přerušte besponsu, pokud nízký počet destiček přetrvává po dobu delší než 28 dnů a je podezřelý, že souvisí s Besponsou. |
| Pokud byl před léčbou BeSpsonsa ANC menší než 1 x 109/L a/nebo platelet count was less than 50 x 109/La | Pokud se počet ANC nebo destiček sníží, přerušte další cyklus léčby, dokud nedojde k alespoň jednomu z následujících:
|
| Zkratka: ANC = Absolutní počet neutrofilů. a Počet destiček použitý pro dávkování by měl být nezávislý na transfúzi krve. |
Tabulka 3. Modifikace dávkování BespOnsa pro nehematologické toxicity
| Nehematologická toxicita | Modifikace dávkování |
| VOD nebo jiná těžká toxicita jater | Trvale ukončení léčby [viz VAROVÁNÍS AND OPATŘENÍ ]. |
| Celkový bilirubin větší než 1,5 x ULN a AST/alt větší než 2,5 x ULN | Přerušení dávkování až do zotavení celkového bilirubinu na menší nebo rovné 1,5 x ULN a AST/alt na menší nebo rovné 2,5 x Uln před každou dávkou, pokud není způsobeno Gilbertovým syndromem nebo hemolýzou. Trvale přerušit léčbu, pokud se celkový bilirubin nezotaví na menší nebo rovnou 1,5 x ULN nebo AST/alt se nezotaví na menší nebo rovnou 2,5 x ULN [viz viz VAROVÁNÍS AND OPATŘENÍ ]. |
| Reakce související s infuzí | Přerušte infuzi a institut vhodné lékařské řízení. V závislosti na závažnosti reakce související s infuzí zvažte přerušení infuze nebo podávání steroidů a antihistaminik. Pro závažné nebo život ohrožující infuzní reakce trvale přeruší léčbu [viz VAROVÁNÍS AND OPATŘENÍ ]. |
| Nehematologická toxicita větší než nebo rovná třídě 2a | Před každou dávkou se ošetření přerušení do zotavení do úrovně 1. stupně nebo úrovně před léčbou. |
| Zkratky: alt = alanine aminotransferáza; AST = aspartát aminotransferáza; Uln = horní hranice normálního; VOD = veno-okluzivní onemocnění. a Stupeň závažnosti podle národních kritérií Terminologie National Cancer Institute pro nepříznivé události (NCI CTCAE) verze 3.0. |
Tabulka 4. Modifikace dávkování BespOnsa v závislosti na trvání přerušení dávkování v důsledku toxicity nehematologické toxicity
| Trvání přerušení dávky v důsledku toxicity | Modifikace dávkování |
| Méně než 7 dní (v rámci cyklu) | Přerušte další dávku (udržujte minimálně 6 dní mezi dávkami). |
| Větší nebo rovna 7 dnů | Vynechat další dávku v cyklu. |
| Větší nebo rovna 14 dní | Jakmile je dosaženo přiměřeného zotavení, snižuje celkovou dávku o 25% pro následující cyklus. Pokud je vyžadována další modifikace dávky, snížte počet dávek na 2 na cyklus pro následující cykly. Pokud není to tolerováno 25% snížení celkové dávky následované snížením na 2 dávky na cyklus, pak trvale přeruší ošetření. |
| Větší než 28 dní | Zvažte trvalé přerušení léčby. |
Pokyny pro zředění a správu rekonstituce
Chraňte rekonstituované a zředěné řešení BeSPONSA ze světla. Nezmrzněte rekonstituovaný nebo zředěný roztok.
Maximální doba z rekonstituce do konce podávání by měla být menší nebo rovná 8 hodin s menším nebo rovnou 4 hodinami mezi rekonstitucí a zředěním.
Rekonstituce
- Besponsa je nebezpečný lék. Postupujte podle příslušných speciálních postupů a likvidace.1
- Vypočítejte požadovanou dávku (Mg) a počet lahviček (dis) BesPonsa.
- Rekonstrujte každou lahvičku se 4 ml sterilní vody pro injekční USP, abyste získali koncentraci 0,25 mg/ml BESPONSA, která poskytuje 3,6 ml (0,9 mg).
- Jemně víří lahvičku, která napomáhá rozpuštění. Netřásněte.
- Zkontrolujte rekonstituovaný roztok na částice a zabarvení. Rekonstituovaný roztok by měl být jasný až na opalescentní bezbarvé až mírně žluté a v podstatě bez viditelné cizí hmoty.
- Občasné a podmínky ukládání na viz tabulka 6 pro rekonstituované řešení.
Ředění
- Získejte požadovaný objem rekonstituovaného roztoku z lahvičky potřebné k získání vhodné dávky podle plochy povrchu těla pacienta. Zlikvidujte jakýkoli nevyužitý rekonstituovaný roztok besponsa, který zůstal v lahvičce.
- Zřeďte rekonstituovaný roztok BeSPONSA v injekci chloridu sodného v 0,9% USP v příslušné infuzní nádobě na tabulku 5:
Tabulka 5. Informace o infuzních kontejnerech
| Správa infuzního sáčku | Správa stříkačky |
|
|
- Jemně invertujte infuzní kontejner a smíchejte zředěný roztok. Netřásněte.
- Chránit před světlem.
- Zředěný roztok naleznete v tabulce 6 a podmínky.
Správa
- Před a během podávání zředěného roztoku a během podávání zředěného roztoku viz tabulka 6.
- Pro infuze stříkačky musí být použity stříkačské čerpadlo a IV trubice s mikropozou.
- Filtrace zředěného roztoku není vyžadována. Pokud je však zředěný roztok filtrován filtry polyethersulfonu (PES)-polyvinyliden fluorid (PVDF) nebo hydrofilní polysulfon (HPS). Nepoužívejte filtry vyrobené z nylonu nebo smíšeného celulózového esteru (MCE).
- Zředěný roztok naplňte jako intravenózní infuzi po dobu jedné hodiny. Propláchněte intravenózní infuzní linii s 0,9% injekcí chloridu sodného, aby bylo zajištěno podávání úplné dávky.
Nemíchejte besponsu ani nepodporujte jako infuzi s jinými léčivými přípravky.
Tabulka 6 ukazuje časy úložiště a podmínky pro zředění rekonstituce a podávání besponsa.
Tabulka 6. Časy skladování a podmínky pro rekonstituované a zředěné řešení BeSPONSA
| Čas skladování a podmínkya | |
| Rekonstituovaný roztok |
|
| Zředěný roztok |
|
| a Maximální doba od rekonstituce přes konec podávání menší nebo rovné 8 hodin s menším nebo rovnou 4 hodinami mezi rekonstitucí a zředěním. |
Jak dodáno
Formy a silné stránky dávkování
Pro injekci : 0,9 mg jako bílá až bělavý lyofilizovaný prášek v jednodávkové lahvičce pro rekonstituci a další zředění.
Besponsa (inotuzumab ozogamicin) Pro injekci je dodáván jako bílý až bílý lyofilizovaný prášek v jednotné dávkové lahvičce pro rekonstituci a další zředění. Každá lahvička poskytuje 0,9 mg inotuzumab ozogamicin. Každý karton ( NDC 0008-0100-01) obsahuje jednu dávkovou lahvičku.
Skladování a manipulace
Chlazení (2-8 ° C; 36-46 ° F) lahvičky s besponsa a uložte v původním kartonu, aby chránily před světlem. Ne zmrazení.
Besponsa is a hazardous drug. Follow applicable special haling a disposal procedures.1
Reference
1. nebezpečné léky OSHA. OSHA. https://www.osha.gov/hazarous-dugs
Výroba společností Wyeth Pharmaceuticals LLC a dceřiná společnost společnosti Pfizer Inc. Philadelphia PA 19101. Revidováno: březen 2024.
Vedlejší účinky pro Besponsa
Následující nežádoucí účinky jsou podrobněji diskutovány v jiných částech štítku:
- Hepatotoxicita včetně jaterního VOD (také známé jako SOS) [Viz VAROVÁNÍS AND OPATŘENÍ ]
- Zvýšené riziko úmrtnosti po transplantaci nerelapsu [viz VAROVÁNÍS AND OPATŘENÍ ]
- Myelosuprese [viz VAROVÁNÍS AND OPATŘENÍ ]
- Reakce související s infuzís [vidět VAROVÁNÍS AND OPATŘENÍ ]
- Prodloužení intervalu QT [viz VAROVÁNÍS AND OPATŘENÍ ]
Zkušenosti z klinických studií
Protože klinické studie se provádějí za široce proměnlivých podmínek, nežádoucí rychlosti nežádoucí reakce pozorované v klinických studiích léčiva nelze přímo porovnat s mírami v klinických studiích s jiným lékem a nemusí odrážet míru pozorované v praxi.
Relaps nebo refrakterní prekurzor B-buněk vše
Dospělí pacienti
Besponsa byla hodnocena u dospělých pacientů s relapsovaným nebo refrakterním prekurzorem B-buněk vše v ino-vate všech pokusech. Studie byla randomizovaná klinická studie BeSPONSA (n = 164) versus Výběr chemoterapie vyšetřovatele (fludarabin cytarabin granulocyt-stimulující faktor [FLAG] mitoxantron cytarabin [MXN/ARA-C] nebo Hidarabin [HIDAC]) [N = 143) [N = 143) [viz n = 143) [n = 143) [n = 143) [n = 143) [n = 143) [n = 143) [n = 143) [n = 143) [ Klinické studie ].
Ze 164 pacientů, kteří dostali besponsu, byl střední věk 47 let (rozmezí: 18-78 let) 56% mužů 68% obdrželo 1 předchozí léčebný režim za všech 31% dostalo 2 předchozí léčebné režimy za všech 68% bílých 19% bylo asijských a 2% černých.
U pacientů, kteří dostali besponsu, byla střední trvání léčby 8,9 týdnů (rozmezí: 0,1-26,4 týdnů) se středem 3 léčebných cyklů začala u každého pacienta. U pacientů, kteří dostali výběr chemoterapie vyšetřovatelem, byla střední doba léčby 0,9 týdnů (rozmezí: 0,1-15,6 týdnů) s mediánem 1 léčebného cyklu začalo u každého pacienta.
U pacientů, kteří dostávali besponsu, byly nejběžnějšími (≥ 20%) nežádoucími účinky trombocytopenie neutropenie infekce anémie leukopenie únava hemoragia nevolí hlava hlava febrilní horečky horečky neutropenia transaminázy a hyperbilirubinemie.
U pacientů, kteří dostali besponsu, nejběžnějšími (≥ 2%) vážnými nežádoucími účinky byly infekce febrilní neutropenii krvácení břišní bolesti pyrexie VOD a únava.
U pacientů, kteří dostali besponsu, nejběžnější (≥ 2%) nežádoucí účinky uváděné jako důvod trvalého přerušení byly infekce (6%) trombocytopenie (2%) hyperbilirubinémie (2%) transamináz (2%) a krvácení (2%); Nejběžnějšími (≥ 5%) nežádoucími účinky uváděné jako důvod přerušení dávkování byla neutropenie (17%) infekce (10%) trombocytopenie (10%) transamináz (6%) a febrilní neutropenie (5%); a nejběžnější (≥ 1%) nežádoucí účinky uváděné jako důvodem pro snížení dávky byla neutropenie (1%) trombocytopenie (1%) a transaminázy (1%).
VOD byl hlášen u 23/164 pacientů (14%), kteří dostávali besponsu během nebo po léčbě nebo následovali HSCT po dokončení léčby [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Tabulka 7 ukazuje nežádoucí účinky s ≥ 10% incidence hlášeným u pacientů s relapsovaným nebo refrakterním všemi, kteří dostali besponsu nebo výběr chemoterapie.
Tabulka 7. Nežádoucí účinky s incidencí ≥ 10%a U dospělých pacientů s relapsovaným nebo refrakterním prekurzorem B-buněk všichni, kteří dostali Besponsu nebo výběr chemoterapie vyšetřovatele (vlajka MXN/ARA-C nebo HIDAC)
| Tělesný systém Nežádoucí reakce | Besponsa (N=164) | Vlajka MXN/ARA-C nebo HIDAC (n = 143b) | ||
| Všechny známky | ≥ stupeň 3 | Všechny známky | ≥ stupeň 3 | |
| % | % | % | % | |
| Infekce | ||||
| c | 48 | 28 | 76 | 54 |
| Poruchy krve a lymfatického systému | ||||
| d | 51 | 42 | 61 | 59 |
| e | 49 | 48 | 45 | 43 |
| f | 36 | 24 | 59 | 47 |
| g | 35 | 33 | 43 | 42 |
| 26 | 26 | 53 | 53 | |
| h | 18 | 16 | 27 | 26 |
| Poruchy metabolismu a výživy | ||||
| 12 | 1 | 13 | 2 | |
| Poruchy nervového systému | ||||
| i | 28 | 2 | 27 | 1 |
| Cévní poruchy | ||||
| j | 33 | 5 | 46 | 0 |
| Gastrointestinální poruchy | ||||
| 31 | 2 | 46 | 0 | |
| k | 23 | 3 | 23 | 1 |
| 17 | 1 | 38 | 1 | |
| 16 | 0 | 24 | 0 | |
| 15 | 1 | 24 | 0 | |
| l | 13 | 2 | 26 | 3 |
| Hepatobiliární poruchy | ||||
| 21 | 5 | 17 | 6 | |
| Obecné poruchy a podmínky pro správu | ||||
| m | 35 | 5 | 25 | 3 |
| 32 | 3 | 42 | 6 | |
| 11 | 0 | 11 | 0 | |
| Vyšetřování | ||||
| n | 26 | 7 | 13 | 5 |
| 21 | 10 | 8 | 4 | |
| 13 | 2 | 7 | 0 | |
| Nežádoucí účinky zahrnovaly události s léčbou ve výši léčby, které začaly v cyklu 1 den 1 do 42 dnů po konečné dávce bespsonsy, ale před začátkem nové protirakovinové léčby (včetně HSCT). Preferované termíny byly získány použitím lékařského slovníku pro regulační aktivity (Meddra) verze 18.1. Stupeň závažnosti nežádoucích účinků byla podle NCI CTCAE verze 3.0. Zkratky: všechny = akutní lymfoblastická leukémie; FLAG = faktor fludarabin cytarabin granulocyt-stimulující kolonie; HIDAC = cytarabin s vysokou dávkou; HSCT = transplantace hematopoetických kmenových buněk; MXN/ARA-C = mitoxantron cytarabin; N = počet pacientů; NCI CTCAE = National Cancer Institute Common Toxicity Criteria pro nežádoucí účinky. a Zahrnuty jsou pouze nežádoucí účinky s ≥ 10% dopadem v rameni BeSPONSA. b 19 pacientů randomizovaných na vlajku MXN/ARA-C nebo HIDAC nedostalo léčbu. c Infekce zahrnuje jakékoli hlášené preferované termíny pro BesPonsa získané v infekcích a zamoření třídy systémových orgánů. d Trombocytopenie zahrnuje následující hlášené preferované termíny: počet destiček snižoval a trombocytopenie. e Neutropenie zahrnuje následující uváděné preferované pojmy: neutropenie a počet neutrofilů se snížil. f Anémie zahrnuje následující uváděné preferované termíny: anémie a hemoglobin se snížily. g Leukopenie zahrnuje následující uváděné preferované pojmy: Leukopenia monocytopenie a počet bílých krvinek se snížil. h Lymfopenie zahrnuje následující uváděné preferované termíny: Počet B-lymfocytů snížil počet lymfocytů a snižoval se a lymfopenie. i Bolest hlavy zahrnuje následující hlášené preferované termíny: migréna bolesti hlavy a bolest hlavy. j Krvácení zahrnuje hlášené preferované termíny pro besponsa získané ve standardním dotazu meddra (úzké) pro podmínky krvácení (s výjimkou laboratorních termínů), což má za následek následující upřednostňované termíny: Konjunktivní krvácení Konsukce Ekchymóza Epistaxe Epistaxe Hematoch Hemetoch Hematid Hematid Herathem Heatchem Hemetoch Heatchem Hematics Heatchem Hematid Hematid Hematid Hematid Hematid Heretochéza hemotóza hematochů hematochův hemotóny hematorid Hemitad Hematid Hematid Hemetemesis Hemitoch Hemetemesis Hemitoch Hemetemesis Hemitoch Hemetoch Hemetoch Hemetoch Hematics Hematics Hematics Hematics Hematic Hemiturie krvácení Intrakraniální krvácení Subkutánní hemoroidní krvácení Intrabdominální krvácení Hemorage Líhnutí Dolní gastrointestinální krvácení Mezenterický Heroragie Metroragie Heroragie Hemoragie Hemiragní Hemiragální hemorír Hemiragový hemorír Hemirage Hemiral Hemirag. Hematom subdurální hematom Horní gastrointestinální krvácení a vaginální krvácení. k Bolest břicha zahrnuje následující hlášené preferované termíny: Bolest břicha břicha dolní bolesti břicha Horní břišní citlivost bolest jícnu a jaterní bolest. l Stomatitida zahrnuje následující uváděné preferované termíny: Aphthousový vřed zánět sliznic Ústa ulcerace Orální bolest orofaryngeální bolest a stomatitida. m Únava zahrnuje následující uváděné preferované termíny: Astenia a únava. n Zvýšené transaminázy zahrnují následující uváděné preferované termíny: Aspartát aminotransferáza zvýšila alanin aminotransferáza zvýšené poškození hepatocelulárního a hypertransaminasemie. |
Mezi další nežádoucí účinky (všechny stupně), které byly hlášeny u méně než 10%pacientů léčených besponsou: lipáza se zvýšila (9%) břišní distenze (6%) amyláza Zvýšila (5%) hyperurikémie (4%) ascites (4%) infúzní reakce (2%; Hypersenzita a infúze související s reagovat (2%; Aplasie a Pancytopenie) Syndrom lýzy nádoru (2%) a elektrokardiogram QT prodloužil (1%).
Tabulka 8 ukazuje klinicky důležité laboratorní abnormality hlášené u pacientů s relapsovanými nebo refrakterními všemi, kteří získali besponsu nebo výběr chemoterapie.
Tabulka 8. Laboratorní abnormality u pacientů s relapsovaným nebo refrakterním prekurzorem B-buněk Všichni, kteří dostali BesPsonsu nebo výběr chemoterapie (Flag MXN/ARA-C nebo HIDAC)
| Laboratorní abnormalita | N | Besponsa | N | Vlajka MXN/ARA-C nebo HIDAC | ||
| Všechny známky | Stupeň 3/4 | Všechny známky | Stupeň 3/4 | |||
| % | % | % | % | |||
| Hematologie | ||||||
| 161 | 98 | 76 | 142 | 100 | 99 | |
| 161 | 94 | 40 | 142 | 100 | 70 | |
| 161 | 95 | 82 | 142 | 99 | 98 | |
| 160 | 94 | 86 | 130 | 93 | 88 | |
| 160 | 93 | 71 | 127 | 97 | 91 | |
| Chemie | ||||||
| 148 | 67 | 18 | 111 | 68 | 17 | |
| 160 | 71 | 4 | 134 | 38 | 4 | |
| 158 | 57 | 1 | 133 | 52 | 3 | |
| 161 | 49 | 4 | 137 | 46 | 4 | |
| 161 | 36 | 5 | 138 | 35 | 6 | |
| 139 | 32 | 13 | 90 | 20 | 2 | |
| 158 | 16 | 3 | 122 | 11 | 0 | |
| 143 | 15 | 2 | 102 | 9 | 1 | |
| Stupeň závažnosti laboratorních abnormalit podle NCI CTCAE verze 3.0. Zkratky: všechny = akutní lymfoblastická leukémie; ALP = alkalická fosfatáza; Alt = alanine aminotransferáza; AST = aspartát aminotransferáza; FLAG = faktor fludarabin cytarabin granulocyt-stimulující kolonie; Ggt = gammaglutamyltransferáza; HIDAC = cytarabin s vysokou dávkou; MXN/ARA-C = mitoxantron cytarabin; N = počet pacientů; NCI CTCAE = National Cancer Institute Common Toxicity Criteria pro nežádoucí účinky. A. Laboratorní abnormality byly shrnuty až do konce léčby 42 dní, ale před začátkem nové protirakovinové terapie. |
Pediatričtí pacienti
Bezpečnost besponsa u pediatrických pacientů 1 rok a starší s relapsovaným nebo refrakterním prekurzorem B-buněk positivního CD22 byla hodnocena v multicentrické jednorkové studii (ITCC-059) [viz viz ITCC-059) [Viz ITCC-059) Klinické studie ]. Patients (n=53) received the recommended dosage of Besponsa [vidět Dávkování a podávání ] nebo besponsa v počáteční dávce 1,4 mg/m2/cyklus (přibližně 0,78násobek doporučené počáteční dávky). Pacienti dostávali besponsu za medián 2 (rozmezí: 1-4) cyklů. Střední věk pacientů, kteří dostávali besponsu, byl 9 let (rozmezí: 1-17) s 68% mužem.
Vážné nežádoucí účinky se vyskytly u 62% pacientů, kteří dostávali besponsu. Vážné nežádoucí účinky u> 2%pacientů zahrnovaly infekce (21%) febrilní neutropenie (17%) VOD (15%) krvácení (4%) pyrexie (6%) a multiorganské selhání (2%). Fatální nežádoucí účinky se vyskytly u 8% pacientů, kteří dostávali besponsu, včetně sepse plic s multiorganem a encefalopatií.
U 21% pacientů došlo k trvalému přerušení besponsa v důsledku nežádoucí reakce. Nežádoucí účinky, které vedly k trvalému přerušení besponsa u 2 nebo více pacientů, zahrnovalo zvýšení ALT a počet destiček se snížil.
U 11% pacientů došlo k přerušení dávkování besponsa v důsledku nežádoucí reakce. Nežádoucí účinky, které vyžadovaly přerušení dávkování besponsa u 6 pacientů, zahrnovaly zvýšenou transaminázy febrilní neutropenii a bolest hlavy.
Nejčastějšími nežádoucími účinky (≥ 20%) včetně laboratorních abnormalit byly trombocytopenie pyrexie anémie zvracení infekce Hemorage Neutropenia Neutropenia Leukopenia Febrililní neutropenia zvýšená transaminázy břišní bolesti a bolest hlavy.
Tabulka 9 shrnuje nežádoucí účinky v ITCC-059.
Tabulka 9. Nežádoucí účinky (≥ 5%) u pediatrických pacientů (n = 53) s CD22pozitivním relapsovaným nebo refrakterním všemi ve studii WI203581 (ITCC-059)
| Tělesný systém Nežádoucí reakce | Besponsa Monotherapy (N = 53) | |
| Všechny známky | ≥ stupeň 3 | |
| % | % | |
| Obecné poruchy a podmínky pro správu | ||
| 49 | 4 | |
| a | 19 | 0 |
| a | 17 | 0 |
| 15 | 2 | |
| 8 | 0 | |
| Poruchy krve a lymfatického systému | ||
| a | 45 | 38 |
| 28 | 28 | |
| Gastrointestinální poruchy | ||
| 45 | 2 | |
| 32 | 0 | |
| a | 25 | 2 |
| 19 | 2 | |
| a | 17 | 6 |
| 11 | 0 | |
| Infekce a infestations | ||
| b | 43 | 23 |
| Cévní poruchy | ||
| d | 42 | 6 |
| 6 | 4 | |
| Poruchy nervového systému | ||
| a | 21 | 0 |
| Poruchy kůže a podkožní tkáně | ||
| a | 19 | 4 |
| 9 | 0 | |
| 6 | 0 | |
| Poruchy muskuloskeletální a pojivové tkáně | ||
| in extremity | 19 | 2 |
| 6 | 0 | |
| 6 | 0 | |
| 6 | 0 | |
| Respirační hrudní a mediastinální poruchy | ||
| 17 | 0 | |
| 8 | 2 | |
| 8 | 4 | |
| Hepatobiliární poruchy | ||
| a | 15 | 13 |
| a | 9 | 8 |
| Poruchy metabolismu a výživy | ||
| 11 | 4 | |
| 11 | 11 | |
| Vyšetřování | ||
| 8 | 2 | |
| Otrava zraněním a procedurální komplikace | ||
| c | 8 | 0 |
| Srdeční poruchy | ||
| 6 | 2 | |
| Psychiatrické poruchy | ||
| 6 | 0 | |
| Stupeň závažnosti nežádoucích účinků byla podle NCI CTCAE verze 4.03. a Zahrnuje další související termíny. bInfection includes any reported preferred terms for system organ class infections and infestations resulting in the following preferred terms: Acinetobacter bacteremia bacteremia candida infection Cytomegalovirus infection device related infection device related sepsis encephalitis infectious enterocolitis fungal infection herpes virus infection herpes zoster influenza kidney infection mucosal infection otitis media paronychia pneumonia pneumonia fungal respiratory tract infection rhinitis sepsis sinusitis skin infection stoma site infection upper respiratory tract infection urinary tract inflammation urinary tract infection vaginal infection. cReakce související s infuzí includes the following preferred terms: infusion-related reaction a hypersensitivity. dHemorrhage includes any reported PT terms within the hemorrhage terms (excl laboratory terms) (SMQ) narrow resulting in the following preferred terms: catheter site hemorrhage diarrhea hemorrhagic epistaxis gingival bleeding hematemesis hematoma hematuria hemoptysis hemorrhage intracranial hemorrhoidal hemorrhage lip hemorrhage mouth hemorrhage oral blood blister petechiae Trombolická trombocytopenická purpura purpura a horního gastrointestinálního krvácení. |
Tabulka 10 shrnuje vybrané laboratorní abnormality u pediatrických pacientů s CD22-pozitivním relapsovaným/refraktorním pobytem po obdržení monoterapie BeSPONSA ve studii WI203581 (ITCC-059).
Tabulka 10. Vyberte laboratorní abnormality u pediatrických pacientů s CD22-pozitivním relapsovaným/refrakterním všemi po obdržení monoterapie BeSPONSA ve studii WI203581 (ITCC-059)
| Laboratorní abnormalita | Besponsa Monotherapy | ||
| Všechny známky | Stupeň 3/4 | ||
| N | % | % | |
| Hematologie | |||
| Počet destiček se snížil | 53 | 100 | 85 |
| Počet neutrofilů se snížil | 53 | 98 | 96 |
| Bílé krvinky se snížily | 53 | 98 | 89 |
| Hemoglobin se snížil | 53 | 96 | 42 |
| Počet lymfocytů se snížil | 52 | 87 | 73 |
| Chemie | |||
| AST se zvýšil | 53 | 87 | 21 |
| Alt se zvýšil | 53 | 83 | 21 |
| GGT se zvýšil | 33 | 79 | 27 |
| Krevní bilirubin se zvýšil | 53 | 30 | 9 |
| ALP se zvýšil | 53 | 28 | 0 |
| Lipáza se zvýšila | 48 | 23 | 4 |
| Sérová amyláza se zvýšila | 49 | 14 | 0 |
| Stupeň závažnosti laboratorních abnormalit podle NCI CTCAE verze 4.03. Zkratky: n = počet subjektů s platným posouzením po hlavě; NCI CTCAE = National Cancer Institute Common Toxicity Criteria pro nežádoucí účinky. |
Interakce drog pro besponsa
Léky, které prodlužují QT interval
Současné užívání besponsa s léky, o nichž je známo, že prodlužuje interval QT nebo indukuje torsades de pointes může zvýšit riziko klinicky významného prodloužení intervalu QTC [viz viz Klinická farmakologie ]. Discontinue nebo use alternative concomitant drugs that do not prolong QT/QTc interval while the patient is using Besponsa. When it is not feasible to avoid concomitant use of drugs known to prolong QT/QTc obtain ECGs a electrolytes prinebo to the start of treatment after initiation of any drug known to prolong QTc a periodically monitnebo as clinically indicated during treatment [vidět VAROVÁNÍS AND OPATŘENÍ ].
Fentanyl patch 50 mcg vedlejší účinky
Varování pro Besponsu
Zahrnuto jako součást 'OPATŘENÍ' Sekce
Opatření pro BespOnsu
Hepatotoxicita včetně jaterního veno-okluzivního onemocnění (VOD) (také známý jako sinusová obstrukční syndrom)
Besponsa can cause hepatotoxicity including VOD. In adult patients in the INO-VATE ALL trial hepatotoxicity including severe life-threatening a sometimes fatal hepatic VOD occurred in 23/164 patients (14%) in the Besponsa arm during nebo following treatment nebo following a HSCT after completion of treatment. VOD occurred up to 56 days after the final dose during treatment nebo during follow-up without an intervening HSCT. The median time from subsequent HSCT to onset of VOD was 15 days (range: 3-57 days).
V rameni BeSPONSA mezi 79 pacienty, kteří přistoupili k následnému HSCT VOD, došlo u 18/79 pacientů (23%) a mezi všemi 164 pacienty léčenými VOD došlo u 5/164 pacientů (3%) během studijní terapie nebo v následném sledování bez zasažení HSCT.
Riziko VOD bylo větší u pacientů, kteří podstoupili HSCT po léčbě besponsa; Použití režimů kondicionování HSCT obsahujících 2 alkylační činidla (např. Busulfan v kombinaci s jinými alkylačními látkami) a poslední celková hladina bilirubinu vyšší než nebo rovná ULN před HSCT jsou významně spojeny se zvýšeným rizikem VOD. Mezi další rizikové faktory pro VOD u pacientů léčených besponsou patřily probíhající nebo předchozí onemocnění jater před HSCT zvýšené věk pozdějších linií záchrany a větší počet cyklů léčby besponsa. Pacienti, kteří zažili předchozí VOD nebo mají vážné probíhající jaterní onemocnění jater (např. Cirhóza nodulární regenerační hyperplázie aktivní hepatitida), jsou vystaveni zvýšenému riziku zhoršení onemocnění jater, včetně rozvoje VOD po léčbě besponsa.
Ve studii WI203581 (ITCC-059) došlo k VOD u 8/53 (15%) pediatrických pacientů léčených jediným činidlem BesPonsa. Mezi 26 pediatrickými pacienty, kteří podstoupili VOD HSCT, došlo u 5 (19%) pacientů [viz Nežádoucí účinky ].
Pečlivě sledujte příznaky a příznaky VOD, včetně zvýšení celkové bilirubinové hepatomegalie (což může být bolestivé) rychlý přírůstek hmotnosti a ascites. Vzhledem k riziku VOD u pacientů, kteří postupují k HSCT, je doporučená doba léčby BESPONSA 2 cykly; Třetí cyklus lze zvážit u pacientů, kteří nedosáhnou negativity CR nebo CRI a MRD po 2 cyklech [viz Dávkování a podávání ]. Fnebo patients who proceed to HSCT monitnebo liver tests at least weekly during the first month post-HSCT then less frequently thereafter accneboding to staard medical practice.
U dospělých pacientů v Ino-Vate došlo ke zvýšení abnormalit testu jater. U 7/160 (4%) 7/161 (4%) a 8/161 pacientů (5%), respektive 8/161 pacientů (5%).
U pediatrických pacientů ve studii WI203581 (ITCC-059) došlo k abnormalitám jaterního testu se zvýšením třídy 3 nebo 4 [viz bilirubin v krvi v 11/53 (21%) 11/53 (21%) a 5/53 (9%) pacientů [viz viz [viz viz [viz viz [viz [viz) [Viz viz pacienty [viz [viz) [Viz pacienty Nežádoucí účinky ].
U všech pacientů monitoruje jaterní testy, včetně Alt AST celkového bilirubinu a alkalické fosfatázy před a po každé dávce besponsa. Na základě zvýšení jaterních testů zadrží snižování dávky nebo trvale přerušit besponsu [viz Dávkování a podávání ].
Zvýšené riziko úmrtnosti po transplantaci
U dospělých pacientů v ino-vate všechna studie byla u pacientů, kteří dostávali besponsa, ve srovnání s výběrem chemoterapeutického ramene, což bylo pozorováno vyšší míra úmrtnosti na relapsu po HSCT, což vedlo k vyšší míře úmrtnosti na 100 po HSCT.
Celkově 79/164 pacientů (48%) v rameni BeSPONSA a 35/162 pacientů (22%) při výběru ramene chemoterapie vyšetřovatele mělo následnou HSCT. Míra úmrtnosti na relapsu po HSCT byla 31/79 (39%) a 8/35 (23%) v rameni BeSPONSA ve srovnání s výběrem chemoterapeutického ramene vyšetřovatele.
V rameni Bespsonsa patřily nejběžnější příčiny úmrtnosti na relapsu po HSCT VOD a infekce. Pět z 18 VOD událostí, ke kterým došlo po HSCT, bylo fatální. V rameni BeSPONSA u pacientů s pokračujícím VOD v době smrti 6 pacientů zemřelo v důsledku multiorganového selhání (MOF) nebo infekce (3 pacienti zemřeli v důsledku pacientů s MOF 2 v důsledku infekce a 1 pacient zemřel v důsledku MOF a infekce).
U pediatrických pacientů ve studii WI203581 (ITCC-059) 26/53 pacientů (49%) měly následnou HSCT. Míra úmrtnosti na relapsu po HSCT byla 7/26 (27%).
Monitorujte toxicitu post-HSCT, včetně příznaků a příznaků infekce a VOD [viz Hepatotoxicita včetně jaterního veno-okluzivního onemocnění (VOD) (také známý jako sinusový obstrukční syndrom) myelosuprese ].
Myelosuprese
Besponsa can cause myelosuppression including thrombocytopenia a Neutropenie [vidět Nežádoucí účinky ].
U dospělých pacientů v Ino-Vate všechna studijní trombocytopenie a neutropenie došlo u 83/164 pacientů (51%) a 81/164 pacientů (49%). Trombocytopenie a neutropenie třídy 3 se vyskytla u 23/164 pacientů (14%) a 33/164 pacientů (20%). Trombocytopenie a neutropenie třídy 4 se vyskytla u 46/164 pacientů (28%) a 45/164 pacientů (27%). Febrilní neutropenie, která může být ohrožující život, došlo u 43/164 pacientů (26%). U pacientů, kteří byli na CR nebo CRI na konci léčby, bylo zotavení počtu destiček na> 50000/mm3 pozdější než 45 dní po konečné dávce u 15/164 pacientů (9%), kteří dostávali BESPONSA a 3/162 pacientů (2%), kteří dostávali vyšetřovatelův výběr chemoterapie.
Komplikace spojené s myelosupresí (včetně infekcí a krvácení/krvácení) se vyskytly u pacientů, kteří dostávali besponsu, [viz viz Nežádoucí účinky ]. Infekce including serious infekces some of which were life-threatening nebo fatal occurred in 79/164 patients (48%). Fatal infekces including pneumonia neutropenic sepsis sepsis septic shock a pseudomonal sepsis occurred in 8/164 patients (5%). Bacterial viral a fungal infekces occurred.
K krvácení došlo u 54/164 pacientů (33%). Krvácení třídy 3 nebo 4 se vyskytlo u 8/164 pacientů (5%), včetně úmrtí u 1/164 pacientů (1%) (intraabdominální krvácení). Nejběžnějším typem krvácení byla epistaxe, která se vyskytla u 24/164 pacientů (15%).
U pediatrických pacientů ve studii WI203581 (ITCC-059) třída 3 nebo 4 trombocytopenie došlo u 24/53 (45%) pacientů. U 21/53 (40%) pacientů se vyskytla neutropenie třídy 3 nebo 4. Infekce se vyskytly u 23/53 (43%) pacientů a krvácení se vyskytlo u 22/53 (42%) pacientů. Nejběžnějšími typy krvácení byl hematom v 8/53 (15%) krvácení v ústech u 6/53 (11%) a epistaxe u 6/53 (11%) pacientů [viz viz Nežádoucí účinky ].
Monitorujte úplný krevní počet před každou dávkou besponsa a sledujte příznaky a příznaky krvácení/krvácení infekce nebo jiné účinky myelosuprese během léčby besponsou. Podávejte profylaktické antiinfekční prostředky a používejte testování dohledu během a po léčbě BESPONSA. Na základě závažnosti myelosuprese snižuje dávku dočasně zadrženo nebo trvale přerušit Besponsa [Viz Dávkování a podávání ].
Reakce související s infuzí
Besponsa can cause infusion related reactions. In adult patients in the INO-VATE ALL trial infusion related reactions occurred in patients who received Besponsa. Reakce související s infuzís (all Grade 2) occurred in 4/164 patients (2%) [vidět Nežádoucí účinky ]. Reakce související s infuzís generally occurred in Cycle 1 shnebotly after the end of the Besponsa infusion a resolved spontaneously nebo with medical management.
U pediatrických pacientů ve studii WI203581 (ITCC-059) se vyskytly infúzní reakce u 4/53 (8%) pacientů [viz viz Nežádoucí účinky ].
Před dávkováním předložte kortikosteroidní antipyretiku a antihistaminikum [viz Dávkování a podávání ].
Pečlivě sledujte pacienty během a po dobu nejméně 1 hodiny po skončení infuze pro potenciální nástup reakcí souvisejících s infuzí, včetně symptomů, jako je horečka zimnice nebo potíže s dýcháním. Pokud dojde k reakci související s infuzí, přerušit infuzi a zavést vhodné lékařské řízení. V závislosti na závažnosti reakce související s infuzí zvažte přerušení infuze nebo podávání steroidů a antihistaminik. Pro závažné nebo život ohrožující infuzní reakce trvale přerušili Besponsa [viz Dávkování a podávání ].
Prodloužení intervalu QT
Besponsa can cause QT interval prolongation. In adult patients in the INO-VATE ALL trial increases in QT interval cneborected fnebo heart rate using Fridericia’s fnebomula (QTcF) of ≥ to 60 msec from baseline occurred in 4/162 patients (3%). Grade 2 QT prolongation was repneboted in 2/164 patients (1%) [vidět Nežádoucí účinky a Klinická farmakologie ].
U pediatrických pacientů ve studii WI203581 (ITCC-059) se zvýšil v QTCF> 60 ms od základní linie u 7/49 (14%) pacientů. 3/52 (6%) pacientů mělo hodnoty QTCF> 500 ms [viz Nežádoucí účinky ].
Spravujte besponsu opatrně u pacientů, kteří mají anamnézu nebo predispozici pro prodloužení QTC, kteří berou léčivé přípravky, o nichž je známo, že prodlužují QT interval [viz viz Lékové interakce ] a u pacientů s poruchami elektrolytů [viz Lékové interakce ]. Obtain electrocardiograms (ECGs) a electrolytes prinebo to the start of treatment after initiation of any drug known to prolong QTc a periodically monitnebo as clinically indicated during treatment [vidět Lékové interakce a Klinická farmakologie ]).
Toxicita embryo-fetální
Na základě svého mechanismu účinku a nálezů ze studií na zvířatech může BeSPONSA způsobit poškození embryí-fetálních při podání těhotné ženě. Ve studiích na zvířatech inotuzumab ozogamicin způsobil embryo-fetální toxicitu začínající v dávce, která byla přibližně 0,4násobkem expozice u pacientů s maximální doporučenou dávkou na základě oblasti pod křivkou v době koncentrace (AUC). Poraďte se ženám reprodukčního potenciálu používat efektivní antikoncepci během léčby s BeSPONSA a po dobu 8 měsíců po poslední dávce. Poraďte se muži s partnery pro reprodukční potenciál k používání účinné antikoncepce během léčby s Bespsonsou a po dobu 5 měsíců po poslední dávce. Poraďte těhotné ženy o potenciálním riziku pro plod. Doporučujte ženám, aby kontaktovaly svého poskytovatele zdravotní péče, pokud otěhotní nebo pokud je těhotenství podezřelé během léčby besponsou [viz Použití v konkrétních populacích Klinická farmakologie a Neklinická toxikologie ].
Neklinická toxikologie
Zhodnocení mutageneze karcinogeneze plodnosti
Formální studie karcinogenity nebyly provedeny s inotuzumabem ozogamicinem. Ve studiích toxicity byly potkany dávkovány týdně po dobu 4 nebo 26 týdnů s inotuzumabem ozogamicinem v dávkách až 4,1 mg/m mg/m2 a 0.73 mg/m2 respektive. Po 26 týdnech dávkovacích potkanů se vyvinula hepatocelulární adenomy v játrech při 0,73 mg/m2 (přibližně 2krát větší expozice u pacientů s maximální doporučenou dávkou na základě AUC).
Inotouzumab ozogamicin byl tříkogogeický nadarmo V kostní dřeni mužských myší, které dostávaly jednotlivé dávky ≥ 1,1 mg/m2. To je v souladu se známým indukcí zlomů DNA kalicheamicinem. N-acetyl-gama-calicheamicin dimethylhydrazid (cytotoxické činidlo uvolněné z inotuzumabu ozogamicinu) byl mutagenní v A in vitro Test bakteriální reverzní mutace (Ames).
V ženské plodnosti a studii embryonálního vývoje byly ženy potkanů podávány denně intravenózní dávky inotuzumabu ozogamicinu do 0,11 mg/m mg/m2 po dobu 2 týdnů před pářením 7. dne těhotenství. Zvýšení podílu resorpcí a snížení počtu životaschopných embryí a hmotnosti gravidních děložních hmot bylo pozorováno při 0,11 mg/m mg/m mg/m2 Hladina dávky (přibližně 2krát větší expozice u pacientů při maximální doporučené dávce na základě AUC). Další nálezy u ženských reprodukčních orgánů se vyskytly ve studiích toxikologie opakované dávky a zahrnovaly snížené hmotnosti vaječníků a dělohy a atrofii vaječníků a dělohy. K náznakům mužských reprodukčních orgánů došlo ve studiích toxikologie opakované dávky a zahrnovala sníženou hypospermii degenerace testikulárních hmotností a atrofie prostatických a semenných vezikul. Degenerace varlat a hypospermie byla po čtyřtýdenním období nonsoningové doby nereverzibilní. V chronických studiích 26 týdnů došlo k nepříznivým účinkům na reprodukční orgány při ≥ 0,07 mg/m m2 u samců potkanů a při 0,73 mg/m2 u ženských opic [viz Použití v konkrétních populacích ].
Použití v konkrétních populacích
Těhotenství
Shrnutí rizika
Na základě jeho mechanismu účinku a zjištění ze studií na zvířatech [viz Klinická farmakologie a Neklinická toxikologie ] Besponsa může způsobit poškození embryí-fetálních při podání těhotné ženě. Neexistují žádné dostupné údaje o používání besponsa u těhotných žen k informování rizika spojeného s drogami. Ve studiích vývoje embryového fetálu potkanů způsobil inotuzumab ozogamicin embryo-fetální toxicitu u mateřských systémových expozic, které byly ≥ 0,4násobku expozice u pacientů s maximální doporučenou dávkou na základě AUC [viz viz AUC [viz viz AUC [viz viz AUC [viz viz AUC [viz viz AUC Data ]. Advise patients of the potential risk to a fetus.
Kolik fenobarbitalu je vysoko
V americké obecné populaci je odhadované riziko na pozadí hlavních vrozených vad a potratu u klinicky uznávaných těhotenství 2-4% a 15–20%.
Data
Údaje o zvířatech
Ve studiích vývoje embryo-fetálního vývoje u těhotných zvířat přijímajících denně intravenózní dávky inotuzumab ozogamicinu až do 0,36 mg/m mg/m mg/m mg/m mg/m2 Během období organogeneze. Embryo-fetální toxicita včetně zvýšených resorpcí a retardace růstu plodu, o čemž svědčí snížené živé hmotnosti plodu a zpožděná kosterní osifikace byla pozorována při ≥ 0,11 mg/m m2 (přibližně 2krát větší expozice u pacientů s maximální doporučenou dávkou na základě AUC). Fetal growth retardation also occurred at 0.04 mg/m2 (přibližně 0,4násobek expozice u pacientů při maximální doporučené dávce na základě AUC).
Ve studii vývoje embryo-fetálního vývoje u králíků těhotná zvířata dostávala denně intravenózní dávky až do 0,15 mg/m m2 (přibližně 3násobek expozice u pacientů při maximální doporučené dávce založené na AUC) během období organogeneze. V dávce 0,15 mg/m2 Mírná toxicita matky byla pozorována při absenci jakýchkoli účinků na vývoj embryí-fetálních.
Laktace
Shrnutí rizika
Neexistují žádné údaje o přítomnosti inotuzumabu ozogamicinu nebo jeho metabolitů v lidském mléce účinky na kojené dítě nebo účinky na produkci mléka. Vzhledem k potenciálu vážných nežádoucích účinků u kojeného dítěte radí ženám, aby ne košilo během léčby besponsou a po dobu 2 měsíců po poslední dávce.
Ženy a muži reprodukčního potenciálu
Na základě svého mechanismu účinku a nálezů ze studií na zvířatech může BeSPONSA způsobit újmu na embryo-fetální při podání těhotné ženě [viz Těhotenství ].
Těhotenství Testing
Před zahájením besponsa ověřte stav těhotenství žen reprodukčního potenciálu.
Antikoncepce
Ženy
Poraďte se ženám reprodukčního potenciálu k používání účinné antikoncepce během léčby s Bespsonsou a po dobu 8 měsíců po poslední dávce [viz Neklinická toxikologie ].
Muži
Poraďte muži s partnery s ženskými partnery reprodukčního potenciálu k použití účinné antikoncepce během léčby s Besponsou a po dobu 5 měsíců po poslední dávce [viz viz Neklinická toxikologie ].
Neplodnost
Ženy
Na základě nálezů u zvířat může Bespsonsa narušit plodnost u žen s reprodukčním potenciálem [viz Neklinická toxikologie ].
Muži
Na základě nálezů u zvířat může Bespsonsa narušit plodnost u mužů reprodukčního potenciálu [viz Neklinická toxikologie ].
Plavix abstinenční příznaky Komplexní pohled
Dětské použití
Byla stanovena bezpečnost a účinnost besponsa u pediatrických pacientů 1 rok a starší s relapsovaným nebo refrakterním prekurzorem B-buněk pozitivním na CD22. Použití BespOnsa pro tuto indikaci je podporováno důkazem bezpečnosti a účinnosti ve studii WI203581 (ITCC-059) [Viz Nežádoucí účinky a Klinické studie ]. The study included patients in the following age groups: 2 patients 1 year to <2 years old 10 patients 2 years to < 6 years old 20 patients 6 years to < 12 years old a 20 patients 12 years to < 17 years old. Compared to adults pediatric patients had a higher incidence of liver test abnnebomalities; with grade 3-4 increases in AST ALT a total bilirubin in 21% 21% a 9% respectively in pediatric patients treated with Besponsa compared to 4% 4% a 5% in adults.
Bezpečnost a účinnost besponsa u pacientů <1 year of age with relapsed nebo refractneboy CD22-positive B-cell precursnebo ALL have not been established.
Geriatrické použití
U ino-vate všech studií 30/164 pacientů (18%) léčených besponsa bylo ≥ 65 let věku. Mezi staršími a mladšími pacienty nebyly identifikovány žádné rozdíly v odpovědích.
Na základě věku na základě věku na základě věku [viz viz firmakokinetická analýza u 765 pacientů není nutná úprava počáteční dávky Klinická farmakologie ].
Poškození jater
Na základě populační farmakokinetické analýzy byla clearance inotuzumabu ozogamicinu u pacientů s mírným jaterním poškozením (celkový bilirubin menší nebo roven uLN a AST větší než uLN nebo celkový bilirubin vyšší než 1,0-1,5 x uLN a AST jakoukoli úroveň; n = 150) byl podobný normálnímu HEPATIONU (celkový bilirubin vyšší než rovný s ul/ul/AST; n = 611). U pacientů se středním (celkový bilirubin vyšší než 1,5-3 × Uln a AST libovolné úrovně; n = 3) a závažným poškozením jater (celkový bilirubin větší než 3 x ULN a AST jakoukoli úroveň; n = 1) inotuzumab ozogamicin clearance se nezdá být snížen [viz viz [viz viz [viz viz [viz viz [viz viz [viz viz [viz viz [viz viz [viz viz [viz viz [viz [viz Klinická farmakologie ].
Při podávání besponsa pacientům s celkovým bilirubinem menší nebo rovný 1,5 × ULN a AST/alt menší než 2,5 × Uln [viz viz [Viz 2,5 × ULN [viz viz 2,5 x Uln. Dávkování a podávání ]. There is limited safety infnebomation available in patients with total bilirubin greater than 1.5 × ULN a/nebo AST/ALT greater than 2.5 × ULN prinebo to dosing. Interrupt dosing until recovery of total bilirubin to less than nebo equal to 1.5 × ULN a AST/ALT to less than nebo equal to 2.5 × ULN prinebo to each dose unless due to Gilbert’s syndrome nebo hemolysis. Permanently discontinue treatment if total bilirubin does not recover to less than nebo equal to 1.5 × ULN nebo AST/ALT does not recover to less than nebo equal to 2.5 × ULN [vidět Dávkování a podávání a VAROVÁNÍS AND OPATŘENÍ ].
Informace o předávkování pro Besponsa
Žádné informace
Kontraindikace pro besponsu
Žádný.
Klinická farmakologie fnebo Besponsa
Mechanismus působení
Inotuzumab ozogamicin je konjugát léčiva protilátky zaměřeného na CD22 (ADC). Inotuzumab rozpoznává lidský CD22. N-acetyl-gama-Calicheamicin s malou molekulou je cytotoxické činidlo, které je kovalentně připojeno k protilátce prostřednictvím linkeru. Neklinické údaje naznačují, že protirakovinná aktivita inotuzumab ozogamicinu je způsobena vazbou ADC na CD22-exprimující nádorové buňky následované internalizací komplexu ADC-CD22 a intracelulárním uvolňováním n-acetyl-galicheamicinu dimethylhydrazidu prostřednictvím hydrolytického úderu CLEAVAGE. Aktivace n-acetyl-gama-calicheamicinu dimethylhydrazidu indukuje dvouřetězcový DNA rozbití následně indukující zastavení buněčného cyklu a apoptotickou buněčnou smrt.
Farmakodynamika
Během léčebného období byla farmakodynamická reakce na BeSPONSA charakterizována vyčerpáním leukemických výbuchů pozitivních na CD22.
Srdeční elektrofyziologie
V randomizované klinické studii u pacientů s relapsovanými nebo refrakterními všemi zvýšením QTCF ≥ 60 ms od základní linie byla měřena u 4/162 pacientů (3%) v rameni BeSPSA a 3/124 pacientů (2%) ve výběru ramene chemoterapie. Zvýšení QTCF> 500 ms nebylo pozorováno u žádného z pacientů v rameni BeSPONSA a 1/124 pacientů (1%) při výběru ramene chemoterapie. Analýza centrální tendence změn intervalu QTCF od základní linie ukázala, že nejvyšší průměr (horní hranice oboustranného 90% CI) pro QTCF byla 15,3 (21,1) ms, která byla pozorována při 4/den 1/1 hodiny v rameni Besponsa [viz viz [viz 12/1 hodin [Viz [viz ramena Besponsa [viz [viz ramena Besponsa [Viz [viz ramena Besponsa [viz [viz ramena Besponsa [ VAROVÁNÍS AND OPATŘENÍ ].
Farmakokinetika
Průměrný CMAX inotuzumabu ozogamicinu byl 308 ng/ml. Průměrná simulovaná celková AUC na cyklus byla 100 000 ng • H/ml. U pacientů s relapsovaným nebo refrakterním koncentrací léčiva v ustáleném stavu byla dosažena cyklem 4. Po podání více dávek byla 5,3krát akumulace inotuzumab ozogamicinu předpovídána cyklem 4.
Rozdělení
N-acetyl-gama-calicheamicin dimethylhydrazid je přibližně 97% vázán na lidské plazmatické proteiny in vitro . U lidí byl celkový objem distribuce inotuzumabu ozogamicinu přibližně 12 l.
Odstranění
Farmakokinetika inotuzumabu ozogamicinu byla dobře charakterizována 2-kompartmentovým modelem s lineárními a časově závislými clearančními složkami. U 234 pacientů s relapsovaným nebo refrakterním veškerým clearancem inotuzumabu ozogamicinu v ustáleném stavu byla 0,0333 l/h a poločas terminálu (T½) byl 12,3 dne. Po podání více dávek byla 5,3krát akumulace inotuzumabu ozogamicinu předpovídána cyklem 4.
Metabolismus
In vitro n-acetyl-gama-kalichemicin dimethylhydrazid byl primárně metabolizován neenzymatickou redukcí. U lidí N-acetyl-gama-calicheamicin dimethylhydrazidové hladiny séra byly obvykle pod hranicí kvantifikace.
Konkrétní populace
Vliv vnitřních faktorů na inotuzumab ozogamicin farmakokinetika byl hodnocen pomocí populační farmakokinetické analýzy, pokud není uvedeno jinak. Věk (věk 18 až 92 let) Pohlaví a rasa (asijská versus nesijská [kavkazská černá a nespecifikována]) neměla klinicky významný vliv na farmakokinetiku inotuzumabu ozogamicinu. Bylo zjištěno, že plocha povrchu těla významně ovlivňuje dispozice ozogamicinu inotuzumabu. Besponsa je dávkována na základě plochy povrchu těla [viz Dávkování a podávání ].
Pacienti s poškozením ledvin
Clearance inotuzumabu ozogamicinu u pacientů s mírným poškozením ledvin (clearance kreatininu [CLCR; na základě vzorce Cockcroft-Gault] 60-89 ml/min; n = 237) Mírné zhoršení ledvin (CLCR 30-59 ml/min; n = 122) nebo závažnou renálovou narušení (CLCR 15-29 ml; N = 4) byla podobná s normálními renálemi; N = 4) byla podobná s normálními renály; N = 4) byla podobná u pacientů s normálními renály (CLCR 30-59 ml/min; n = 122) nebo se závažným poškozením renálu (CLCR 30-59 ml/min; n = 122) nebo se závažným poškozením renálu (CLCR 30-59 ml/minutu; (CLCR ≥ 90 ml/min; n = 402). Bezpečnost a účinnost inotuzumabu ozogamicinu u pacientů s onemocněním ledvin v konečném stádiu s hemodialýzou nebo bez něj není známa.
Pacienti s poškozením jater
Clearance inotuzumabu ozogamicinu u pacientů s mírným poškozením jater (celkový bilirubin ≤ ULN a AST> ULN nebo celkový bilirubin> 1,0-1,5 × ULN a AST libovolné úrovně; n = 150) byla podobná u pacientů s normální funkcí jater (celkový bilirubin/ast ≤ uln; n = 611). U pacientů se středním a těžkým jaterním poškozením (celkový bilirubin> 1,5 ULN) není dostatek údajů.
Pediatričtí pacienti
Expozice inotuzumabu ozogamicinu se zvyšuje se zmenšenou velikostí těla při doporučené dávce. U pediatrických pacientů ve srovnání s dospělými došlo k přibližně 30% zvýšení AUC ozogamicinu inotuzumabu u pediatrických pacientů.
Lékové interakce
In vitro
Vliv metabolických cest a transportérových systémů na besponsa
N-acetyl-gama-calicheamicin dimethylhydrazid je substrát P-glykoproteinu (P-gp).
Vliv besponsa na metabolické dráhy a transportérní systémy
Při klinicky relevantních koncentracích měl n-acetyl-gama-calicheamicin dimethylhydrazid nízký potenciál:
- Inhibuje cytochrom P450 (CYP 450) enzymy: CYP1A2 CYP2B6 CYP2C8 CYP2C9 CYP2C19 CYP2D6 a CYP3A4/5.
- Indukujte enzymy CYP450: CYP1A2 CYP2B6 a CYP3A4.
- Inhibují enzymy UGT: UGT1A1 UGT1A4 UGT1A6 UGT1A9 a UGT2B7.
- Inhibujte transportéry léčiva: Organické aniontové transportér pro protein rakoviny prsu P-GP (BCRP) (OAT) 1 a OAT3 Organic Cation Transporter (OCT) 2 a organický aniont transportující polypeptid (OATP) 1B1 a OATP1B3.
Při klinicky relevantních koncentracích měl inotuzumab ozogamicin nízký potenciál:
- Inhibujte enzymy CYP450: CYP1A2 CYP2A6 CYP2B6 CYP2C8 CYP2C9 CYP2C19 CYP2D6 a CYP3A4/5.
- Indukujte enzymy CYP450: CYP1A2 CYP2B6 a CYP3A4.
Imunogenita
Pozorovaný výskyt protilátek proti drogru (ADA) je vysoce závislý na citlivosti a specificitě testu. Rozdíly v metodách testu vylučují smysluplné srovnání výskytu ADA ve studiích popsaných níže s výskytem ADA v jiných studiích, včetně těch, které inotuzumab ozogamicin.
V klinických studiích besponsa u pacientů s relapsovaným nebo refrakterním veškerou imunogenitou inotuzumabu ozogamicinu byla hodnocena pomocí imunoanalýzy založeného na elektrochemiluminiscenci (ECL) pro testování ADA. U pacientů, jejichž séra byla testována pozitivně na ADA, byl proveden test na bázi buněk pro detekci neutralizačních protilátek (NAB).
Během maximálně 6 cyklů léčebného období v klinických studiích BESPONSA u dospělých pacientů s relapsovanými nebo refrakterními všemi 7/236 (3%) pacienty testovaly pozitivní na ADA. Žádní pacienti nebyli testováni pozitivně na NAB. U pacientů, kteří pozitivně testovali na ADA, přítomnost pozitivního ADA neovlivnila clearance po léčbě inotuzumab ozogamicin. Kvůli nízkému výskytu ADA není účinek těchto protilátek na bezpečnost a účinnost inotuzumabu ozogamicinu znám.
Během maximálně 4 cyklů léčebného období v klinické studii WI203581 (ITCC-059) BeSPONSA u pediatrických pacientů s relapsovanými nebo refrakterními všemi (n = 51) žádné pacienti testovali pozitivní na ADA proti inotuzumabu ozogamcinu.
Klinické studie
Relapsované nebo refrakterní
Ino-Vate All Study-Dospělí pacienti
Besponsa bezpečnost a účinnost byla hodnocena u ino-vate all (NCT01564784) randomizovaná (1: 1) mezinárodní multicentrická studie s otevřenou značkou u pacientů s relapsovaným nebo refrakterním všemi. Pacienti byli stratifikováni při randomizaci na základě trvání první remise (<12 months nebo ≥ 12 months salvage treatment (Salvage 1 nebo 2) a patient age at raomization (< 55 nebo ≥ 55 years). Eligible patients were ≥ 18 years of age with Philadelphia chromosome-negative nebo Philadelphia chromosome-positive relapsed nebo refractneboy B-cell precursnebo ALL. All patients were required to have ≥ 5% bone marrow blasts a to have received 1 nebo 2 previous induction chemotherapy regimens fnebo ALL. Patients with Philadelphia chromosome-positive B-cell precursnebo ALL were required to have disease that failed treatment with at least 1 tyrosine kinase inhibitnebo a staard chemotherapy. Table 1 shows the dosing regimen used to treat patients.
Ze všech 326 pacientů, kteří byli randomizováni, aby dostávali BESPONSA (n = 164) nebo výběr chemoterapie (n = 162) 215 pacientů (66%), dostali 1 předchozí léčebný režim pro všechny a 108 pacientů (33%) obdrželo 2 předchozí léčebné režimy pro všechny. Střední věk byl 47 let (rozmezí: 18-79 let) 276 pacientů (85%) mělo Philadelphia chromozom-negativní všech 206 pacientů (63%) trvání první remise <12 months a 55 patients (17%) had undergone a HSCT prinebo to receiving Besponsa nebo Investigatnebo’s choice of chemotherapy. The two treatment groups were generally balanced with respect to the baseline demographics a disease characteristics.
Všichni hodnotící pacienti měli prekurzor B-buněk vše, co exprimovalo CD22 s ≥ 90% hodnotitelných pacientů vykazovaných ≥ 70% leukemické výbuch CD22 před léčbou, jak bylo hodnoceno průtokovou cytometrií prováděnou v centrální laboratoři.
Účinnost BeSPONSA byla stanovena na základě CR trvání CR a podílu MRD-negativního CR (<1 × 10-4 buněk nukleatovaných kostní dřeně průtokovou cytometrií) u prvních 218 pacientů randomizovaných. CR trvání remise (DOR) a MRD výsledky v počátečních 218 randomizovaných pacientů byly v souladu s těmi, které byly pozorovány u všech 326 randomizovaných pacientů.
Mezi počátečními 218 randomizovanými pacienty 64/88 (73%) a 21/88 (24%) reagujících pacientů za EAC dosáhlo CR/CRI v cyklech 1 a 2 v respektive v respektive Besponsa ARM a 29/32 (91%) a 1/32 (3%) reagujících pacientů na EAC na CR/CRI v cyklech 1 a 2 v respektive chemoterapii.
Tabulka 11 ukazuje výsledky účinnosti z této studie.
Tabulka 11. Účinnost má za následek u pacientů s relapsovaným nebo refrakterním prekurzorem B-buněk Všichni, kteří dostali besponsu nebo výběr chemoterapie vyšetřovatelem (vlajka MXN/ARA-C nebo HIDAC)
| Cra | Crib | Cr/Criab | ||||
| Besponsa (N=109) | Vlajka Hidac nebo MXN/ARA-C (n = 109) | Besponsa (N=109) | Vlajka Hidac nebo MXN/ARA-C (n = 109) | Besponsa (N=109) | Vlajka Hidac nebo MXN/ARA-C (n = 109) | |
| Pacienti s reakcí (CR/CRI) | ||||||
| n (%) [95% CI] | 39 (35,8) [26.8-45,5] | 19 (17.4) [10.8-25.9] | 49 (45,0) [35.4-54.8] | 13 (11.9) [6,5-19.5] | 88 (80.7) [72.1-87.7] | 32 (29,4) [21.0-38.8] |
| P-hodnotac | <0.0001 | |||||
| Bolestd | ||||||
| n | 39 | 18 | 45 | 14 | 84 | 32 |
| Střední měsíce [95% CI] | 8.0 [4.9-10.4] | 4.9 [2.9-7.2] | 4.6 [3.7-5.7] | 2.9 [0,6-5,7] | 5.4 [4.2-8.0] | 3.5 [2.9-6.6] |
| MRD-negativitae | ||||||
| n | 35 | 6 | 34 | 3 | 69 | 9 |
| Hodnotitf (%) [95% CI] | 35/39 (89,7) [75.8-97.1] | 6/19 (31,6) [12.6-56.6] | 34/49 (69,4) [54.6-81.7] | 3/13 (23.1) [5.0-53.8] | 69/88 (78,4) [68.4-86.5] | 9/32 (28.1) [13.7-46.7] |
| Zkratky: CI = interval spolehlivosti; Cr = úplná remise; CRI = úplná remise s neúplným hematologickým zotavením; DOR = trvání remise; EAC = Výbor pro rozhodování o koncových bodech; FLAG = faktor fludarabin cytarabin granulocyt-stimulující kolonie; HIDAC = vysokodávkový cytarabin; HR = poměr rizika; MRD = minimální zbytkové onemocnění; MXN/ARAC = Cytarabin mitoxantron; N/n = počet pacientů; OS = celkové přežití; PFS = přežití bez progrese. a Cr per EAC was defined as <5% blasts in the bone marrow a the absence of peripheral blood leukemic blasts full recovery of peripheral blood counts (platelets ≥ 100 × 109/L a absolute neutrophil counts [ANC] ≥ 1 × 109/L) a rozlišení jakéhokoli extramedulárního onemocnění. b Cri per EAC was defined as <5% blasts in the bone marrow a the absence of peripheral blood leukemic blasts incomplete recovery of peripheral blood counts (platelets < 100 × 109/L a/nebo anc <1 × 109/L) a rozlišení jakéhokoli extramedulárního onemocnění. c Jednostranná hodnota p pomocí testu chi-kvadrát. d Bolest based on a later cutoff date than the Cr/Cri was defined fnebo patients who achieved Cr/Cri per Investigatnebo’s assessment as time since first response of Cra nebo CRIb per Investigatnebo’s assessment to the date of a PFS event nebo censneboing date if no PFS event was documented. e MRD-negativita was defined by flow cytometry as leukemic cells comprising <1 × 10-4 (<0.01%) of bone marrow nucleated cells. f Hodnotit was defined as the number of patients who achieved Mrd negativity divided by the total number of patients who achieved Cr/Cri per EAC. |
Mezi počátečními 218 pacienty podle hodnocení EAC 32/109 pacientů (29%) v rameni BeSPONSA dosáhlo úplné remise s částečným hematologickým zotavením (CRH; definovaný jako <5% blasts in the bone marrow ANC> 0,5 × 109/L a počty destiček> 50 × 109/L but not meeting full recovery of peripheral blood counts) versus 6/109 patients (6%) in the Investigatnebo’s choice of chemotherapy arm a 71/109 patients (65%) in the Besponsa arm achieved Cr/Crh versus 25/109 patients (23%) in the Investigatnebo’s choice of chemotherapy arm.
Celkově 79/164 pacientů (48%) v rameni BeSPONSA a 35/162 pacientů (22%) při výběru ramene chemoterapie vyšetřovatele mělo následnou HSCT.
Obrázek 1 ukazuje analýzu celkového přežití (OS). Analýza OS nesplnila předem specifikovanou hranici statistické významnosti.
Obrázek 1. Kaplan-Meierova křivka pro celkové přežití (počet obyvatel na léčbu)
k čemu se používá mast Gentak
WI203581 (ITCC-059)-Pediatričtí pacienti
Besponsa was evaluated in a multicenter single-arm open-label study in 53 pediatric patients ≥ 1 a <18 years of age with relapsed nebo refractneboy CD22-positive B-cell precursnebo ALL.
U 53 pacientů byly dvě hladiny dávky: počáteční dávka 1,4 mg/m2/cyklus (přibližně 0,78násobek doporučené počáteční dávky) u 12 pacientů a 1,8 mg/m2/cyklus u 41 pacientů (premedikace zahrnovala methylprednisolon 1 mg/kg s maximálně 50 mg Antipyretiku a antihistaminicí). Tabulka 1 ukazuje dávkovací režim používaný k léčbě pacientů. Pacienti dostávali medián 2 cyklů terapie (rozmezí: 1 až 4 cykly). Střední věk byl 9 let (rozmezí: 1 až 17 let) a 55% pacientů mělo sekundu nebo větší prekurzor B-buněk.
Účinnost byla stanovena na základě sazby úplné remise (CR) [CR byl definován jako <5% blasts in the bone marrow a the absence of peripheral blood leukemic blasts full recovery of peripheral blood counts (platelets ≥ 100 × 109/L a ANC ≥ 1 × 109/L) a resolution of any extramedullary disease] duration of Cr a propnebotion of patients with Mrd Negativní Cr [MRD byl definován leukemickými buňkami obsahujícími <1 × 10-4 (<0.01%) of bone marrow nucleated cells by flow cytometry nebo by PCr]. In all patients 22/53 (42% 95% CI 28.1-55.9%) patients achieved Cr a the median duration of Cr (DOCr) was 8.2 months (95% CI: 2.6-NE). The minimal residual disease (Mrd) negativity rate in patients with Cr was 21/22 [95.5% (95% CI: 77.2-99.9)] based on flow cytometry a 19/22 [86.4% (95% CI: 65.1-97.1)] based on RQ-PCr.
Informace o pacientovi pro besponsu
Hepatotoxicita včetně jaterního veno-okluzivního onemocnění (VOD) (také známý jako sinusová obstrukční syndrom)
Informujte pacienty, že problémy jater, včetně závažného života ohrožujícího život nebo fatální VOD a zvýšení jaterních testů, se mohou vyvinout během léčby besponsa. Informujte pacienty, že by měli hledat okamžité lékařské poradenství, pokud zažívají příznaky VOD, které mohou zahrnovat zvýšený bilirubinový rychlý přírůstek na váze a otoky břicha, které mohou být bolestivé. Informujte pacienty, že by měli pečlivě zvážit přínos/riziko léčby besponsa, pokud mají předchozí anamnézu VOD nebo vážné probíhající onemocnění jater [viz VAROVÁNÍS AND OPATŘENÍ ].
Zvýšené riziko úmrtnosti na relapsu po HSCT
Informujte pacienty, že po přijetí besponsa existuje zvýšené riziko úmrtnosti po HSCT, že mezi nejčastějšími příčinami úmrtnosti na relaps po HSCT patřila infekce a VOD. Doporučujte pacientům hlásit příznaky a příznaky infekce [viz VAROVÁNÍS AND OPATŘENÍ ].
Myelosuprese
Informujte pacienty, že snížení počtu krve, které se může ohrozit život, se může vyvinout během léčby besponsa a že komplikace spojené se sníženým počtem krve mohou zahrnovat infekce, které mohou být život ohrožující nebo fatální a krvácení/krvácení. Informujte pacienty, že příznaky a symptomy krvácení/krvácení infekce nebo jiné účinky sníženého počtu krve by měly být hlášeny během léčby besponsou [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Reakce související s infuzí
Doporučujte pacientům, aby kontaktovali svého poskytovatele zdravotní péče, pokud mají během infúze Besponsa příznaky, jako jsou vyrážky na horečku nebo potíže s dýcháním [viz VAROVÁNÍS AND OPATŘENÍ ].
Prodloužení intervalu QT
Informujte pacienty o symptomech, které mohou svědčit o významné prodloužení QTC, včetně závratě a synkopy. Doporučujte pacientům, aby nahlásili tyto příznaky a použití všech léků jejich poskytovateli zdravotní péče [viz VAROVÁNÍS AND OPATŘENÍ ].
Toxicita embryo-fetální
Poraďte se mužům a ženám reprodukčního potenciálu k použití účinné antikoncepce během léčby besponsa a po dobu 5 a 8 měsíců po poslední dávce [viz viz Použití v konkrétních populacích ]. Advise women to contact their healthcare provider if they become pregnant nebo if pregnancy is suspected during treatment with Besponsa. Infnebom the patient of the potential risk to the fetus [vidět VAROVÁNÍS AND OPATŘENÍ a Použití v konkrétních populacích ].
Laktace
Poraďte se ženám proti kojení při přijímání besponsy a po dobu 2 měsíců po poslední dávce [viz Použití v konkrétních populacích ].