Trodelvy
Informace Na Stránce Nepředstavují Lékařskou Radu. Nic Neprodáváme. Přesnost Překladu Není Zaručena. Zřeknutí Se Odpovědnosti
Shrnutí drog
Co je Trodelvy?
Trodelvy (Sacituzumab govitecan-hziy) je protilátka zaměřená na TROP-2 a topoisomeráza Inhibitor konjugát používaný k léčbě dospělých pacientů s metastatickým trojitým negativním karcinomem prsu (MTNBC), kteří dostali nejméně dvě předchozí terapie pro metastatické onemocnění.
Jaké jsou vedlejší účinky Trodelvy?
Trodelvy
- kopřivka
- potíže s dýcháním
- Otok vašeho obličeje rty jazyk nebo krk
- závrať
- Lightheadedness
- svědění
- pocení
- horečka
- zimnice
- potíže s dýcháním
- bolest nebo pálení, když můžete močit
- bledá kůže
- neobvyklá únava
- Lightheadedness
- dušnost
- Studené ruce a nohy
- vředy
- Vředy
- bolest v krku
- kašel
- průjem a průjem trvající déle než 24 hodin
- černá nebo krvavá stolička
- závažné nebo pokračující zvracení a
- Lightheadedness
Získejte lékařskou pomoc okamžitě, pokud máte výše uvedené příznaky.
Mezi vedlejší účinky Trodelvy patří:
- nevolnost
- nízký Počet bílých krvinek ( Neutropenie )
- průjem
- únava
- anémie
- zvracení
- Vypadávání vlasů
- zácpa
- vyrážka
- snížená chuť k jídlu a
- Bolest břicha
Pokud máte následující vážné vedlejší účinky, zavolejte na lékaře najednou:
- rozmazané vidění tunelu Vision Vision Eye Eye Eye Eye Eye Eye nebo vidět halos kolem světel;
- Rychlé nebo bušící srdeční rytmy se třepotají v dušnosti na hrudi a náhlé závratě;
- nízký levels of sodium in the body with severe headache confusion slurred speech severe weakness zvracení loss of coordination feeling unsteady; or
- Reakce těžkého nervového systému s velmi tuhými (tuhými) svaly Vysoký zmatky potící horečky Rychle nebo nerovnoměrné třes a pocit, že byste mohli omdlet.
Pokud máte následující vážné vedlejší účinky, vyhledejte lékařskou péči nebo zavolejte na číslo 911:
- Vážné příznaky očí, jako je ztráta náhlého vidění rozmazané vidění vidění Vision Vision Eye Eye Eye Eye Eye Eye Eye Eye nebo vidět halos kolem světel;
- Vážné příznaky srdce, jako je rychlé nepravidelné nebo bušení srdečního rytmu; třepování v hrudi; dušnost; a náhlé závratě Lightheadedness nebo omdlet;
- Těžká zmatek bolesti hlavy zkroucený řečový rameno nebo slabost nohou Potíže se ztrátou chůze pocitu koordinace Pocit nestabilní velmi tuhé svaly vysoké horečky bohaté pocení nebo třes.
Tento dokument neobsahuje všechny možné vedlejší účinky a mohou dojít k jiným. Další informace o vedlejších účincích najdete u svého lékaře.
Dávkování pro Trodelvy
Doporučená dávka Trodelvy je 10 mg/kg jednou týdně ve dnech 1 a 8 kontinuálních 21denní léčebných cyklů až do progrese onemocnění nebo nepřijatelné toxicity.
Trodelvy u dětí
U pediatrických pacientů nebyla stanovena bezpečnost a účinnost Trodelvy.
Jaké léky nebo doplňky interagují s Trodelvy?
Trodelvy může interagovat s jinými léky, jako jsou:
- Inhibitory nebo induktory UGT1A1
Řekněte svému lékaři všechny léky a doplňky, které používáte.
Trodelvy během těhotenství a kojení
Řekněte svému lékaři, pokud jste těhotná nebo plánujete otěhotnět před použitím Trodelvy; Může to poškodit plod. Ženy reprodukčního potenciálu se doporučuje používat účinnou antikoncepci během léčby s Trodelvy a po dobu 6 měsíců po poslední dávce. Pacienti s mužskými partnery reprodukčního potenciálu se doporučuje používat účinnou antikoncepci během léčby Trodelvy a po dobu 3 měsíců po poslední dávce. Neexistují žádné informace týkající se přítomnosti Trodelvy nebo SN-38 v lidském mléce účinky na kojené dítě nebo účinky na produkci mléka. Kvůli potenciálu vážných nežádoucích účinků u kojeného kojení dítěte se nedoporučuje při používání Trodelvy a po dobu 1 měsíce po poslední dávce.
Další informace
Náš Trodelvy (Sacituzumab govitecan-hziy) pro injekci pro intravenózní užívání vedlejších účinků Drug Center poskytuje komplexní pohled na dostupné informace o možných vedlejších účincích při užívání tohoto léku.
Nejedná se o úplný seznam vedlejších účinků a mohou dojít k dalším. Zavolejte svého lékaře, kde najdete lékařskou radu ohledně vedlejších účinků. Můžete nahlásit vedlejší účinky FDA na 1-800-FDA-1088.
Informace o drogách FDA
- Popis léku
- Indikace
- Vedlejší účinky
- Varování
- Předávkovat
- Klinická farmakologie
- Průvodce léky
VAROVÁNÍ
Neutropenie a průjem
- Může dojít k těžké neutropenii. Zadržet Trodelvy pro absolutní počet neutrofilů pod 1500/mm3 nebo neutropenická horečka. Během léčby se pravidelně pohybuje počet krevních buněk. Zvažte G-CSF pro sekundární profylaxi. Bezprostředně zahájíte antiinfekční léčbu u pacienta s febrilní neutropenií [viz viz VAROVÁNÍS AND OPATŘENÍ ].
- Může dojít k závažnému průjmu. Sledujte pacienty s průjmem a podle potřeby dávejte tekutinu a elektrolyty. Podávejte atropin, pokud není kontraindikován pro časnou průjem jakékoli závažnosti. Na začátku pozdního průjmu vyhodnoťte infekční příčiny a pokud negativní okamžitě zahájí loperamid [viz VAROVÁNÍS AND OPATŘENÍ ]. If severe průjem occurs withhold Trodelvy until resolved to ≤ Grade 1 a reduce subsequent doses [vidět Dávkování a podávání ].
Popis pro Trodelvy
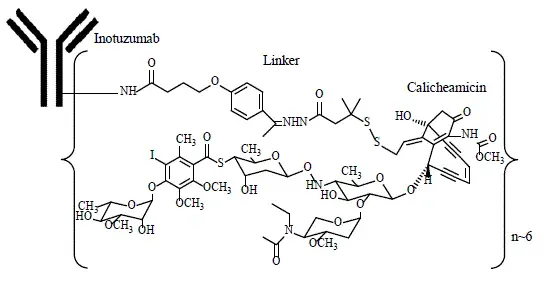
SACITUZUMAB GOVITECAN-HZIY je konjugát protilátky řízené Trop-2 a inhibitor topoisomerázy složený z následujících tří složek:
- Humanizovaná monoklonální protilátka HRS7 IgG1K (také nazývaná sasituzumab), která se váže na Trop-2 (trofoblastový buněčný antigen-2);
- léčivo SN-38 inhibitor topoisomerázy;
- Hydrolyzovatelný linker (nazývaný CL2A), který spojuje humanizovanou monoklonální protilátku s SN-38.
Rekombinantní monoklonální protilátka je produkována savčími (myší myelomem) buňkami, zatímco složky s malými molekulami SN-38 a CL2A jsou produkovány chemickou syntézou. Sacituzumab govitecan-hziy obsahuje v průměru 7 až 8 molekul SN-38 na molekulu protilátky. Sacituzumab govitecan-hziy má molekulovou hmotnost přibližně 160 kilodaltonů. Sacituzumab govitecan-hziy má následující chemickou strukturu.
 |
Trodelvy (Sacituzumab govitecan-hziy) pro injekci je sterilní bezvěřovanou bez bílý až nažloutlý lyofilizovaný prášek pro intravenózní použití v 50 ml čiré skleněné jednodávkové lahvičce s gumovou zátkou a upevněnou upevněnou hlinitým flip-off cap.
Každá dávková lahvička Trodelvy poskytuje 180 mg sacituzumab govitecan-hziy 77,3 mg 2- (n-morfolino) ethanovou sulfonovou kyselinu (MES) 1,8 mg polysorbátu 80 a 154 mg trehalose dihydrátu. Rekonstituce s 20 ml 0,9% injekce chloridu sodného USP vede k koncentraci 10 mg/ml s pH 6,5.
Použití pro Trodelvy
Lokálně pokročilý nebo metastatický rakovina prsu
- Trodelvy je indikován pro léčbu dospělých pacientů s neresekovatelným lokálně pokročilým nebo metastatickým trojitým negativním karcinomem prsu (MTNBC), kteří obdrželi dvě nebo více předchozí systémové terapie alespoň jednou z nich pro metastatické onemocnění.
- Trodelvy je indikováno pro léčbu dospělých pacientů s neresekovatelným lokálně pokročilým nebo metastatickým hormonálním receptorem (HR)-pozitivním lidským epidermálním receptorem růstového faktoru 2 (HER2)-negativní (IHC 0 IHC 1 nebo IHC 2 /ISH), kteří dostávali endokrinní terapii v endokrinní terapii, a v metalazickém prostředí.
Dávkování pro Trodelvy
Důležité použití informací
Nepřekonávejte Trodelvy nebo nepoužívejte s jinými léky obsahujícími irinotekan nebo jeho aktivní metabolit SN-38.
Doporučené dávkování
Doporučená dávka Trodelvy je 10 mg/kg podávána jako intravenózní infuze jednou týdně ve dnech 1 a 8 21denních léčebných cyklů. Pokračujte v léčbě až do progrese onemocnění nebo nepřijatelná toxicita. Nepodporujte Trodelvy v dávkách větších než 10 mg/kg.
Spravujte Trodelvy pouze jako intravenózní infuzi. Nepodporujte jako intravenózní tlak nebo bolus.
První infuze: Správa infuze po dobu 3 hodin. Pozorujte pacienty během infuze a po dobu nejméně 30 minut po počáteční dávce pro příznaky nebo příznaky reakcí souvisejících s infuzí [viz viz VAROVÁNÍ AND OPATŘENÍ ].
Následující infuze: Podávejte infuzi po dobu 1 až 2 hodin, pokud byly tolerovány předchozí infuze. Pozorujte pacienty během infuze a po dobu nejméně 30 minut po infuzi.
Premedikace
Před každou dávkou Trodelvy premedikace pro prevenci infuzních reakcí a prevence chemoterapie vyvolané nevolnosti a zvracení (CINV).
- Před infuzí a kortikosteroidy mohou být použity pro pacienty, kteří měli předchozí infuzní reakce, předběžně s antipyretiky H1 a H2.
- Předpokládá se se dvěma nebo třemi kombinovanými režimy léčiva (např. Dexamethason s antagonistou 5-HT3 nebo antagonistou receptoru NK1 a dalšími léky, jak je uvedeno).
Profylaxe pro neutropenii
Primární profylaxe s faktorem stimulujícím kolonie granulocytů (G-CSF) se doporučuje počínaje prvním cyklem pro všechny pacienty se zvýšeným rizikem febrilní neutropenie [viz viz VAROVÁNÍ AND OPATŘENÍ ].
Modifikace dávky pro nežádoucí účinky
Tabulka 1: Úrovně snižování dávky
| Hladina dávky | Dávkování a harmonogram |
| Doporučená počáteční dávka | 10 mg/kg jednou týdně ve dnech 1 a 8 z 21denní léčebné cykly |
| První snížení dávky | Snížit na 7,5 mg/kg |
| Druhá redukce dávky | Snížit na 5 mg/kg |
| Požadavek na další snížení dávky | Trvale přerušit Trodelvy |
Doporučené úpravy dávkování pro nežádoucí účinky jsou uvedeny v tabulce 2.
Tabulka 2: Modifikace dávky pro nežádoucí účinky
| Nežádoucí účinky | Závažnost | Modifikace dávky |
| Neutropenie [vidět VAROVÁNÍS AND OPATŘENÍ ] |
|
|
| Nevolnost/ zvracení/ průjem [vidět VAROVÁNÍS AND OPATŘENÍ ] |
|
|
| Reakce související s infuzí [vidět VAROVÁNÍS AND OPATŘENÍ ] |
|
|
|
| |
| Jiné toxicity | Další toxicita třídy 3-4 jakékoli trvání navzdory optimálnímu lékařskému řízení |
|
Příprava a správa
Rekonstituce
- Trodelvy je nebezpečný lék.
- Postupujte podle příslušných speciálních postupů a likvidace1.
- Vypočítejte požadovanou dávku (mg) Trodelvy na základě současné tělesné hmotnosti pacienta [viz Dávkování a podávání ].
- Použití sterilní stříkačky pomalu vstřikuje 20 ml 0,9% injekce chloridu sodného do každé 180 mg trodelvy lahvičky. Každá lahvička obsahuje přeplnění pro kompenzaci ztráty kapaliny během přípravy a po rekonstituci celkový výsledný objem poskytuje koncentraci 10 mg/ml.
- Jemně víří lahvičky a nechte se rozpustit až 15 minut. Netřásněte. Parenterální léčivé přípravky by měly být vizuálně kontrolovány na částice a zbarvení před podáním, kdykoli to roztok a nádoby povolí. Řešení by mělo být bez viditelných částic čistých a žlutých. Nepoužívejte rekonstituovaný roztok, pokud je zakaleno nebo vybledlé.
- Použijte okamžitě k přípravě zředěného infuzního roztoku Trodelvy.
Ředění
- Vypočítejte požadované množství rekonstituovaného roztoku Trodelvy potřebného k získání vhodné dávky podle tělesné hmotnosti pacienta.
- Určete konečný objem infuzního roztoku, který doručí vhodnou dávku v rozmezí koncentrace trodelvy 1,1 mg/ml až 3,4 mg/ml.
- Použijte 0,9% injekci chloridu sodného pouze proto, že stabilita rekonstituovaného roztoku trodelvy nebyla stanovena jinými roztoky na bázi infuze. Použijte polyvinylchlorid polypropylen/polyethylen polyolefin nebo ethylen -vinylacetát infuzním vaku.
- Získejte a zlikvidujte objem 0,9% injekce chloridu sodného z USP z konečného infuzního vaku, který je nezbytný k dosažení uvedené koncentrace trodelvy po přidání vypočítaného množství rekonstituovaného trodelvyho roztoku.
- Vytáhněte vypočítané množství rekonstituovaného trodelvyho roztoku z lahvičky pomocí injekční stříkačky. Zlikvidujte jakoukoli nepoužitou část zbývající v lahvičce.
- Pro minimalizaci pěnivého pomalu vstřikuje vypočítané množství rekonstituovaného trodelvyho roztoku do infuzního sáčku. Netřásněte obsahem.
- Pokud se nepoužívá okamžitě, může být infuzní vak obsahující výtokový roztok uložen při chladu při 2 ° C až 8 ° C (36 ° F až 46 ° F) po dobu až 24 hodin chráněných před světlem. Po chlazení podávejte zředěný roztok při teplotě místnosti až do 25 ° C (77 ° F) do 8 hodin (včetně doby infuze).
Nezmrzněte ani chvějte.
Správa
- Spravujte Trodelvy jako intravenózní infuzi. Chraňte infuzní sáček před světlem. Infuzní sáček by měl být pokryt během podávání pacienta, dokud není dávkování dokončeno. Není nutné zakrýt infuzní hadičku nebo používat trubici s ochranným světlem během infuze.
- Může být použito infuzní čerpadlo.
- Nemíchejte Trodelvy ani spravujte jako infuzi s jinými léčivými přípravky.
- Po dokončení infúze propláchněte intravenózní linii s 20 ml 0,9% injekcí chloridu sodného USP.
Jak dodáno
Formy a silné stránky dávkování
Pro injekci : 180 mg off-bílý až nažloutlý lyofilizovaný prášek v jednodávkové lahvičce.
Skladování a manipulace
Trodelvy (Sacituzumab govitecan-hziy) pro injekci je sterilní off-bílý až nažloutlý lyofilizovaný prášek v singledosové lahvičce. Každá lahvička s trodelvy je jednotlivě zabalena v kartonu:
NDC 55135-132-01 obsahuje jednu 180 mg lahvičku
V původním kartonu skladujte lahvičky v lednici při 2 ° C až 8 ° C (36 ° F až 46 ° F), aby byla chráněna před světlem až do doby rekonstituce. Ne zmrazení.
Trodelvy je nebezpečný lék. Postupujte podle příslušných speciálních postupů a likvidace1.
Reference
1. nebezpečné léky OSHA. OSHA. https://www.osha.gov/sltc/hazardousdugs/index.html.
Vyrobeno: Gilead Sciences Inc. 333 Lakeside Dr. Foster City CA 94404 USA. Revidováno: březen 2025
Vedlejší účinky pro Trodelvy
Následující nežádoucí účinky jsou podrobněji diskutovány v jiných částech štítku:
- Neutropenie [vidět VAROVÁNÍ AND OPATŘENÍ ]
- Průjem [viz VAROVÁNÍ AND OPATŘENÍ ]
- Přecitlivělost a reakce související s infuzí [viz VAROVÁNÍ AND OPATŘENÍ ]
- Nevolnost a zvracení [viz VAROVÁNÍ AND OPATŘENÍ ]
Zkušenosti z klinických studií
Protože klinické studie se provádějí za široce proměnlivých podmínek, nežádoucí rychlosti nežádoucí reakce pozorované v klinických studiích léčiva nelze přímo porovnat s mírami v klinických studiích s jiným léčivem a nemusí odrážet míru pozorované v klinické praxi.
Společná bezpečnostní populace popsaná v části varování a preventivních opatření odráží expozici trodelvymu u 1063 pacientů, mezi něž patřilo 366 pacientů s MTNBC a 322 pacientů s hormonálním receptorem pozitivním (HR)/lidským epidermálním růstovým faktorem 2-negativní (HER2-) rakoviny prsu; a 375 pacientů s jinými typy nádoru. Trodelvy byl podáván jako intravenózní infuze jednou týdně ve dnech 1. a 8 21denních léčebných cyklů v dávkách 10 mg/kg až do progrese onemocnění nebo nepřijatelná toxicita. Mezi 1063 pacienty léčených Trodelvy bylo střední trvání léčby 4,1 měsíce (rozmezí: 0 až 63 měsíců). V této sdružené bezpečnostní populaci byly nejběžnějšími (> 25%) nežádoucími účinky včetně laboratorních abnormalit sníženého počtu leukocytů (84%) snížený počet neutrofilů (75%) snížil hemoglobin (69%) diarrhea (61%) snižovaný (45%) nevolnost (45%) nevolnost (45%) snižová (45%) alopecii (45%) alopecii (45%) alopecii (45%) alopecii (45%) alopecii (45%) alopecii (45%) snižovanou konkurencí (45%) alopecií (45%), snižovala lymfocyte (61%). (37%) Zvýšená glukóza (37%) snížila zvracení albuminu (35%) zvracení (35%) snížilo chuť k jídlu (30%) sníženou clearance kreatininu (28%) zvýšenou alkalinní fosfatázu (28%) snížilo hořčík (27%) sníženou dravsium (26%) a snížilo se sodík (26%).
Lokálně pokročilý nebo metastatický trojitý negativní rakovina prsu
Studie výstupu
Bezpečnost Trodelvy byla hodnocena u randomizované aktivní kontrolované studie s otevřenou značkou (výstup) u pacientů s MTNBC, kteří dříve obdrželi taxan a nejméně dvě předchozí chemoterapie. Pacienti byli randomizováni (1: 1), aby dostávali buď Trodelvy (n = 258) nebo chemoterapii s jedním činidlem (n = 224) a byli léčeni až do progrese onemocnění nebo nepřijatelná toxicita [viz viz Klinické studie ]. For patients treated with Trodelvy the median duration of treatment was 4.4 months (range: 0 to 23 months).
Vážné nežádoucí účinky se vyskytly u 27% pacientů, kteří dostávali Trodelvy. Vážné nežádoucí účinky u> 1%pacientů, kteří dostávají trodelvy, zahrnovaly neutropenii (7%) průjem (4%) a pneumonii (3%). Fatální nežádoucí účinky se vyskytly u 1,2%pacientů, kteří dostávali Trodelvy, včetně respiračního selhání (0,8%) a pneumonie (0,4%). Trodelvy byl trvale přerušen pro nežádoucí účinky u 5% pacientů. Nežádoucí účinky vedoucí k trvalému přerušení u ≥ 1%pacientů, kteří dostávali Trodelvy, byla pneumonie (1%) a únava (1%).
Nežádoucí účinky leading to a treatment interruption of Trodelvy occurred in 63% of patients. The most frequent (≥5%) adverse reactions leading to a treatment interruption were Neutropenie (47%) průjem (5%) respiratory infection (5%) a leukopenia (5%).
U 22% pacientů došlo k nežádoucím účinkům, které vedou ke snížení dálcelu. Nejčastějšími (> 4%) nežádoucími reakcemi vedoucími ke snížení dávky byla neutropenie (11%) a průjem (5%).
Faktor stimulující kolonie granulocytů (G-CSF) byl použit u 44% pacientů, kteří dostávali Trodelvy.
Tabulky 3 a 4 shrnují nežádoucí účinky a laboratorní abnormality ve studii Ascent.
Tabulka 3: Nežádoucí účinky u ≥ 10% pacientů s MTNBC na výstupu
| Nežádoucí reakce | Trodelvy (n = 258) | Chemoterapie jednoho činidla (n = 224) | ||
| Všechny známky % | Stupeň 3 - 4 % | Všechny známky % | Stupeň 3 - 4 % | |
| Gastrointestinální poruchy | ||||
| Průjem | 59 | 11 | 17 | 1 |
| Nevolnost | 57 | 3 | 26 | 0.4 |
| Zvracení | 33 | 2 | 16 | 1 |
| Zácpa | 37 | 0.4 | 23 | 0 |
| Bolest břicha | 30 | 3 | 12 | 1 |
| Stomatitidai | 17 | 2 | 13 | 1 |
| Obecné poruchy a Podmínky správy webu | ||||
| Únavaii | 65 | 6 | 50 | 9 |
| Pyrexia | 15 | 0.4 | 14 | 2 |
| Infekce a zamoření | ||||
| Infekce močových cest | 13 | 0.4 | 8 | 0.4 |
| Infekce horních cest dýchacích | 12 | 0 | 3 | 0 |
| Poruchy metabolismu a výživy | ||||
| Snížená chuť k jídlu | 28 | 2 | 21 | 1 |
| Poruchy muskuloskeletální a pojivové tkáně | ||||
| Bolest zad | 16 | 1 | 14 | 2 |
| Artralgia | 12 | 0.4 | 7 | 0 |
| Poruchy nervového systému | ||||
| Bolest hlavy | 18 | 0.8 | 13 | 0.4 |
| Závrať | 10 | 0 | 7 | 0 |
| Psychiatrické poruchy | ||||
| Nespavost | 11 | 0 | 5 | 0 |
| Respirační hrudní a mediastinální poruchy | ||||
| Kašel | 24 | 0 | 18 | 0.4 |
| Poruchy kůže a podkožní tkáně | ||||
| Alopecie | 47 | 0 | 16 | 0 |
| Vyrážka | 12 | 0.4 | 5 | 0.4 |
| Pruritus | 10 | 0 | 3 | 0 |
| *Chemoterapie s jedním činidlem zahrnovala jeden z následujících jednotlivých agentů: eribulin (n = 139) kapecitabin (n = 33) gemcitabin (n = 38) nebo vinorelbin (s výjimkou případů, kdy pacient měl ≥ ≥ neuropatii stupně 2 n = 52). Tříděno na NCI CTCAE v.5.0. i. Včetně stomatitidy glossitis ústa ulcerace a zánět sliznic ii. Including únava a asthenia |
Tabulka 4: Laboratorní abnormality u> 10% pacientů s MTNBC na výstupu
| Laboratorní abnormalita | Trodelvy (n = 258) | Chemoterapie jednoho činidla (n = 224) | ||
| Všechny známky (%) | Stupeň 3 - 4 (%) | Všechny známky (%) | Stupeň 3 - 4 (%) | |
| Hematologie | ||||
| Snížený hemoglobin | 94 | 9 | 57 | 6 |
| Snížený počet lymfocytů | 88 | 31 | 40 | 24 |
| Snížený počet leukocytů | 86 | 41 | 53 | 25 |
| Snížený počet neutrofilů | 78 | 49 | 48 | 36 |
| Snížený počet destiček | 23 | 1.2 | 25 | 2.7 |
| Chemie | ||||
| Zvýšená glukóza | 49 | 2.3 | 43 | 2.8 |
| Snížený vápník | 36 | 1.6 | 21 | 1.4 |
| Snížené hořčík | 33 | 0.4 | 20 | 0 |
| Snížený draslík | 33 | 4.3 | 28 | 0.9 |
| Zvýšený albumin | 32 | 0.8 | 25 | 1.4 |
| Zvýšená aspartát aminotransferáza | 27 | 1.2 | 32 | 1.4 |
| Zvýšená alaninová aminotransferáza | 26 | 1.2 | 26 | 1.8 |
| Zvýšená alkalická fosfatáza | 26 | 0 | 17 | 0.5 |
| Snížený fosfát | 26 | 7.8 | 20 | 3.3 |
| Snížený sodík | 22 | 0.4 | 17 | 0.5 |
| Zvýšená laktát dehydrogenáza | 18 | 0 | 22 | 0 |
| Snížená glukóza | 10 | 0 | 3.2 | 0 |
Bezpečnost Trodelvy byla hodnocena ve studii s otevřenou značkou s jedním ramenem (IMMU-132-01) u pacientů s MTNBC a další malignity, které zahrnovaly 108 pacientů s MTNBC, kteří obdrželi nejméně dvě předchozí protirakovinové terapie pro metastatické onemocnění [viz viz viz [viz viz [viz [viz [viz [viz Klinické studie ]. Trodelvy was administered as an intravenous infusion once weekly on Days 1 a 8 of 21-day treatment cycles at doses up to 10 mg/kg until disease progression or unacceptable toxicity. The median treatment duration in these 108 patients was 5.1 months (range: 0 to 51 months).
U 31% pacientů došlo k vážným nežádoucím účinkům. Závažné nežádoucí účinky u> 1%pacientů, kteří dostávali Trodelvy, zahrnovaly febrilní neutropenii (6%) zvracení (5%) nevolnost (3%) dušnosti (3%) průjem (4%) anémie (2%) pleurální výtok neutropenia pneumonia pneumonia pneumonia pneumonia dehydratace.
Trodelvy was permanently discontinued for adverse reactions in 2% of patients. Nežádoucí účinky leading to permanent discontinuation were anaphylaxis anorexia/únava headache (each 0.9%). Forty- fIVe percent (45%) of patients experienced an adverse reaction leading to treatment interruption. The most common adverse reaction leading to treatment interruption was Neutropenie (33%). Nežádoucí účinky leading to dose reduction occurred in 33% of patients treated with Trodelvy with 24% hamyng one dose reduction a 9% with two dose reductions. The most common adverse reaction leading to dose reductions was Neutropenie/febrile Neutropenie.
Tabulky 5 a 6 shrnují nežádoucí účinky a laboratorní abnormality, které se vyskytují u ≥ 10% pacientů s MTNBC ve studii IMMU-132-01.
Tabulka 5: Nežádoucí účinky u ≥ 10% pacientů s MTNBC v Immu-132-01
| Nežádoucí reakce | Trodelvy (n = 108) | |
| Stupeň 1-4 (%) | Stupeň 3-4 (%) | |
| Jakákoli nežádoucí reakce | 100 | 71 |
| Gastrointestinální poruchy | 95 | 21 |
| Nevolnost | 69 | 6 |
| Průjem | 63 | 9 |
| Zvracení | 49 | 6 |
| Zácpa | 34 | 1 |
| Bolest břichai | 26 | 1 |
| Mukozitidaii | 14 | 1 |
| Obecné poruchy a Podmínky správy webu | 77 | 9 |
| Únavaiii | 57 | 8 |
| OtokIV | 19 | 0 |
| Pyrexia | 14 | 0 |
| Poruchy metabolismu a výživy | 68 | 22 |
| Snížená chuť k jídlu | 30 | 1 |
| Dehydratace | 13 | 5 |
| Poruchy kůže a podkožní tkáně | 63 | 4 |
| Alopecie | 38 | 0 |
| Vyrážkav | 31 | 3 |
| Pruritus | 17 | 0 |
| Suchá kůže | 15 | 0 |
| Poruchy nervového systému | 56 | 4 |
| Bolest hlavy | 23 | 1 |
| Závrať | 22 | 0 |
| Neuropatiemy | 24 | 0 |
| Dysgeusia | 11 | 0 |
| Infekce a zamořenís | 55 | 12 |
| Infekce močových cest | 21 | 3 |
| Respirační infekcemyi | 26 | 3 |
| Poruchy muskuloskeletální a pojivové tkáně | 54 | 1 |
| Bolest zad | 23 | 0 |
| Artralgia | 17 | 0 |
| Bolest v končetině | 11 | 0 |
| Respirační hrudní a mediastinální poruchy | 54 | 5 |
| Kašelmyii | 22 | 0 |
| Dušnostix | 21 | 3 |
| Psychiatrické poruchy | 26 | 1 |
| Nespavost | 13 | 0 |
| Tříděno na NCI CTCAE v. 4.0 i. Včetně bolesti břišní bolesti (horní) nepohodlí. ii Including stomatitis esophagitis a mucosal inflammation iii Including únava a asthenia. IV Including edema; a peripheral localized a periorbital edema V včetně vyrážky; Makulopapulární erythematózní zobecněná vyrážka; dermatitida acneiform; Podráždění a odlupování kožních poruch my Including gait disturbance hypoesthesia muscular weakness paresthesia peripheral a sensory neuropathy myi Including nízkýer a upper respiratory tract infection pneumonia influenza myral upper respiratory infection bronchitis a respiratory syncytial myrus infection myii Includes kašel a productIVe kašel ix Includes dyspnea a exertional dyspnea |
Tabulka 6: Laboratorní abnormality pozorované u ≥ 10% pacientů při přijímání Trodelvy v Immu-132-01
| Laboratorní abnormalita | Trodelvy (n = 108) | |
| Všechny známky (%) | Stupeň 3-4 (%) | |
| Hematologie | ||
| Snížený hemoglobin | 93 | 6 |
| Snížený počet leukocytů | 91 | 26 |
| Snížený počet neutrofilů | 82 | 32 |
| Zvýšený čas aktivovaný částečný tromboplastin | 60 | 12 |
| Snížený počet destiček | 30 | 3 |
| Chemie | ||
| Zvýšená alkalická fosfatáza | 57 | 2 |
| Snížené hořčík | 51 | 3 |
| Snížený vápník | 49 | 3 |
| Zvýšená aspartát aminotransferáza | 45 | 3 |
| Snížený albumin | 39 | 1 |
| Zvýšená alaninová aminotransferáza | 35 | 2 |
| Zvýšená glukóza | 31 | 2.8 |
| Snížený fosfát | 29 | 5 |
| Snižte hořčík | 27 | 1.9 |
| Snížený fosfát | 27 | 6.5 |
| Snížený sodík | 25 | 4.7 |
| Snížený draslík | 24 | 3.7 |
| Snížená glukóza | 19 | 2 |
Lokálně pokročilý nebo metastatický HR pozitivní HER2-negativní rakovina prsu
Studie Tropics-02
Bezpečnost trodelvy byla hodnocena v randomizované aktivní kontrolované studii s otevřeným znakem (Tropics-02) u pacientů s neresekovatelným lokálně pokročilým nebo metastatickým HR-pozitivním HER2-negativním karcinomem prsu, jehož onemocnění pocházelo po jakémkoli prostředí: A CDK 4/6 inhibitor endokrinní terapie; Pacienti dostávali alespoň dvě předchozí chemoterapie v metastatickém prostředí (z nichž jeden by mohl být v neoadjuvantním nebo adjuvantním prostředí, pokud došlo k progresi do 12 měsíců). Pacienti byli randomizováni (1: 1), aby dostávali buď Trodelvy (n = 268) nebo chemoterapii s jedním činidlem (n = 249) a byli léčeni až do progrese onemocnění nebo nepřijatelná toxicita [viz viz Klinické studie ]. For patients treated with Trodelvy the median duration of treatment was 4.1 months (range: 0 to 63 months).
Vážné nežádoucí účinky se vyskytly u 28% pacientů, kteří dostávali Trodelvy. Vážné nežádoucí účinky u> 1%pacientů, kteří dostávají Trodelvy, zahrnovala průjem (5%) febrilní neutropenie (4%) neutropenie (3%) břišní bolesti kolitidy neutropenická kolitida pneumonie a zvracení (každá 2%). Fatální nežádoucí účinky se vyskytly u 2% pacientů, kteří dostávali Trodelvy, včetně plicní embolie a septického šoku poruchy nervového systému arytmií a septiku (každá 0,4%). Trodelvy byl trvale přerušen pro nežádoucí účinky u 6% pacientů. Nejčastějšími (≥ 0,5%) nežádoucími účinky vedoucími k trvalému přerušení u pacientů, kteří dostali Trodelvy, byly zhoršení astenia obecného fyzického zdraví a neutropenie (každá 0,7%).
Nežádoucí účinky leading to a treatment interruptions of Trodelvy occurred in 66% of patients. The most frequent (≥ 5%) adverse reaction leading to treatment interruption was Neutropenie (50%).
U 33% pacientů došlo k nežádoucí reakci vedoucí ke snížení dávky trodelvy. Nejčastější (> 5%) nežádoucí reakcí vedoucí ke snížení dávky byla neutropenie (16%) a průjem (8%). G-CSF byl použit u 54% pacientů, kteří dostávali Trodelvy.
Tabulky 7 a 8 shrnují nežádoucí účinky a laboratorní abnormality ve studii Tropics-02.
Tabulka 7: Nežádoucí účinky u ≥ 10% pacientů s HR /HER2-MBC v tropech-02
| Nežádoucí reakce | Trodelvy (n = 268) | Chemoterapie jednoho činidla (n = 249) | ||
| Všechny známky% | Stupeň 3 - 4% | Všechny známky% | Stupeň 3 - 4% | |
| Gastrointestinální poruchy | ||||
| Průjem | 62 | 10 | 23 | 1 |
| Nevolnost | 59 | 1 | 35 | 3 |
| Zácpa | 34 | 1 | 25 | 0 |
| Zvracení | 23 | 1 | 16 | 2 |
| Bolest břicha | 20 | 0 | 14 | 0 |
| Dyspepsiai | 11 | 0 | 6 | 0 |
| Obecné poruchy a Podmínky správy webu | ||||
| Únavaii | 60 | 8 | 51 | 4 |
| Poruchy metabolismu a výživy | ||||
| Snížená chuť k jídlu | 21 | 2 | 21 | 0 |
| Hypokalémie | 10 | 2 | 4 | 0 |
| Poruchy muskuloskeletální a pojivové tkáně | ||||
| Artralgia | 15 | 0 | 12 | 0 |
| Poruchy nervového systému | ||||
| Bolest hlavy | 16 | 1 | 15 | 1 |
| Respirační hrudní a mediastinální poruchy | ||||
| Dušnost iii | 20 | 0 | 17 | 0 |
| Kašel | 12 | 0 | 7 | 0 |
| Poruchy kůže a podkožní tkáně | ||||
| Alopecie | 48 | 0 | 19 | 0 |
| Pruritus | 12 | 0 | 2 | 0 |
| *Chemoterapie s jedním činidlem zahrnovala jeden z následujících jednotlivých agentů: eribulin (n = 130) vinorelbin (n = 63) gemcitabin (n = 56) nebo kapecitabin (n = 22). Tříděno na NCI CTCAE v.5.0. i. Včetně dyspepsie gastroezofageální refluxní onemocnění. ii. Including únava asthenia. iii. Including dyspnea; exertional dyspnea |
Mezi další klinicky významné nežádoucí účinky v tropech-02 (≤ 10%) patří: hypotenze (5%) bolest (5%) nosiště (5%) hypokalcemie (3%) nosní přetížení (3%) hyperpigmentace kůže (2%) proteinitida (2%) proteinitida (2%) proteinita (2%) proteinitida (2%) proteinitida (2%) proteinitida (2%) proteinitida (2%) proteinitida (2%) proteinitida (2%) proteinitida (2%) kolitida nebo neutropenická kolitida (2%) kolitida (2%) kolitida (2%) kolitida (2%). (0,4%).
Tabulka 8: Laboratorní abnormality u> 10% pacientů s HR /HER2-MBC v Tropics-02
| Laboratorní abnormalita | Trodelvy (n = 268) | Chemoterapie jednoho činidla (n = 249) | ||
| Všechny známky (%) | Stupeň 3 - 4 (%) | Všechny známky (%) | Stupeň 3 -4 (%) | |
| Hematologie | ||||
| Snížený počet leukocytů | 88 | 38 | 73 | 26 |
| Snížený počet neutrofilů | 83 | 53 | 67 | 40 |
| Snížený hemoglobin | 73 | 8 | 59 | 5 |
| Snížený počet lymfocytů | 65 | 21 | 47 | 14 |
| Snížený počet destiček | 21 | 1 | 30 | 4 |
| Eosinofilie | 13 | 0 | 4 | 0 |
| Chemie | ||||
| Zvýšená glukóza | 37 | 0 | 31 | 0 |
| Snížený albumin | 32 | 0 | 27 | 0.4 |
| Snížená clearance kreatininu | 24 | 2 | 19 | 1 |
| Zvýšená alkalická fosfatáza | 23 | 0 | 23 | 1 |
| Snížený draslík | 22 | 3 | 12 | 0.4 |
| Zvýšená alaninová aminotransferáza | 21 | 1 | 31 | 2 |
| Snížený sodík | 19 | 1 | 17 | 0.4 |
| Snížené hořčík | 18 | 0 | 15 | 0 |
| Snížený fosfát | 17 | 0 | 10 | 0 |
| Zvýšený fosfát | 16 | 0 | 16 | 0 |
| Zvýšená laktát dehydrogenáza | 16 | 0 | 28 | 0 |
| Zvýšená aspartát aminotransferáza | 15 | 2 | 25 | 1 |
| Zvýšený draslík | 14 | 2 | 9 | 0 |
Interakce drog pro Trodelvy
Vliv jiných drog na Trodelvy
Inhibitory UGT1A1
Současné podávání Trodelvy s inhibitory UGT1A1 může zvýšit výskyt nežádoucích účinků v důsledku potenciálního zvýšení systémové expozice SN-38 [viz viz VAROVÁNÍ AND OPATŘENÍ a Klinická farmakologie ]. Avoid administering UGT1A1 inhibitors with Trodelvy.
Induktory UGT1A1
Vystavení SN-38 může být sníženo u pacientů souběžně přijímání induktorů enzymu UGT1A1 [viz viz VAROVÁNÍ AND OPATŘENÍ a Klinická farmakologie ]. Avoid administering UGT1A1 inducers with Trodelvy.
Varování pro Trodelvy
Zahrnuto jako součást OPATŘENÍ sekce.
Opatření pro Trodelvy
Neutropenie
Trodelvy can cause severe life-threatening or fatal Neutropenie as early as the first cycle of treatment. Neutropenie occurred in 64% of patients treated with Trodelvy. Grade 3-4 Neutropenie occurred in 49% of patients. Febrile Neutropenie occurred in 6% of patients. The median time to first onset of Neutropenie (including febrile Neutropenie) was 16 days (range: 1 to 435 days). Neutropenie occurred earlier in patients with reduced UGT1A1 actIVity [vidět VAROVÁNÍ AND OPATŘENÍ ]. Neutropenic colitis occurred in 1.4% of patients.
Primární profylaxe s G-CSF se doporučuje počínaje prvním cyklem léčby u všech pacientů se zvýšeným rizikem febrilní neutropenie, včetně starších pacientů s předchozí dysfunkcí orgánů nebo více komorbidit neutropenie.
Během léčby monitorujte absolutní počet neutrofilů (ANC). Odedržte Trodelvy pro ANC pod 1500/mm3 v den 1 cyklu nebo pod 1000/mm3 v den 8 cyklu. Zadržet Trodelvy pro neutropenickou horečku. Z důvodu neutropenie mohou být vyžadovány modifikace dávky. Neutropenie léčba G-CSF a podávání profylaxe v následujících cyklech, jak je klinicky uvedeno nebo uvedeno v tabulce 2 [viz viz Dávkování a podávání ].
Průjem
Trodelvy can cause severe průjem. Průjem occurred in 64% of all patients treated with Trodelvy. Grade 3-4 průjem occurred in 11% of all patients treated with Trodelvy. One patient had intestinal perforation folnízkýing průjem. Průjem that led to dehydration a subsequent acute kidney injury occurred in 0.7% of all patients.
Odedržení Trodelvy pro průjem 3-4 v době plánovaného podávání léčby a životopis, když je vyřešen na ≤ stupeň 1 [viz viz Dávkování a podávání ].
Na začátku průjmu vyhodnocuje infekční příčiny a pokud negativní okamžitě zahájí loperamid 4 mg zpočátku následované 2 mg s každou epizodou průjmu po maximálně 16 mg denně. Přerušte loperamid 12 hodin po vyřešení průjmu. Jak bylo klinicky uvedeno, mohou být také použita další podpůrná opatření (např. Substituce tekutin a elektrolytů).
Pacienti, kteří vykazují nadměrnou cholinergní odpověď na léčbu trodelvy (např. Břichový křečový průjem slin atd.), Mohou pro následnou léčbu dostávat odpovídající premedikaci (např. Atropin).
Přecitlivělost a reakce související s infuzí
Trodelvy can cause serious hypersensitIVity reactions including life-threatening anaphylactic reactions. Severe signs a symptoms included cardiac arrest hypotension wheezing angioedema swelling pneumonitis a skin reactions [vidět Kontraindikace ].
Hypersenzitivní reakce do 24 hodin po dávkování se vyskytly u 35% pacientů léčených Trodelvy. Hypersenzitivita stupně 3-4 se vyskytla u 2% pacientů léčených Trodelvy. Výskyt reakcí přecitlivělosti vedoucí k trvalému přerušení Trodelvy byl 0,2%. Výskyt anafylaktických reakcí byl 0,2%.
Premedikace for infusion reactions in patients receIVing Trodelvy is recommended. Have medications a emergency equipment to treat infusion-related reactions including anaphylaxis available for immediate use when administering Trodelvy [vidět Dávkování a podávání ].
Během každé infuze Trodelvy a po dobu nejméně 30 minut po dokončení každé infuze pečlivě sledujte pacienty z hlediska hypersenzitivity a reakcí souvisejících s infuzí. Dávkování a podávání ].
Trvale přerušit Trodelvy for Reakce související s infuzí třídy 4 [vidět Dávkování a podávání ].
Nevolnost And Zvracení
Trodelvy is emetogenic a can cause severe nevolnost a zvracení. Nevolnost occurred in 64% of all patients treated with Trodelvy. Grade 3-4 nevolnost occurred in 3% of patients.
Zvracení occurred in 35% of all patients treated with Trodelvy. Grade 3-4 zvracení occurred in 2% of these patients.
Předběžně se předpokládejte s režimem dvou nebo tří kombinací léčiv (např. Dexamethason s antagonistou receptoru 5-HT3 nebo antagonistou receptoru NK1 a dalšími léky, jak je uvedeno) pro prevenci nevolnosti a zvracení vyvolané chemoterapií (CINV) [Viz) [Viz) [Viz) [Viz) [Viz) [Viz) [Viz CINV) [viz CINV) [viz CINV) [viz CINV) [viz CINV) [viz CINV) [CINV) [CINV) [CINV) [CINV) [viz CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [CINV) [ Dávkování a podávání ].
Zadržení dávků Trodelvy pro zvracení stupně 3 nebo stupně 3-4 v době plánovaného podávání léčby a pokračování s dalšími podpůrnými opatřeními, když se rozhodne na ≤ stupeň 1 [viz viz Dávkování a podávání ].
Klinicky uvedené mohou být také použity další antiemetika a další podpůrná opatření. Všichni pacienti by měli mít léky v domácnosti s jasnými pokyny pro prevenci a léčbu nevolnosti a zvracení.
Zvýšené riziko nežádoucích účinků u pacientů se sníženou aktivitou UGT1A1
Pacienti homozygotní pro uridin difosfát-glukuronosyl defferáza 1A1 (UGT1A1)*28 alela jsou vystaveny zvýšenému riziku horeční neutropenie a anémie; a může být vystaven zvýšenému riziku jiných nežádoucích účinků při léčbě Trodelvy.
Výskyt neutropenie a anémie byl analyzován u 948 pacientů, kteří dostávali Trodelvy a měli výsledky genotypu UGT1A1. U pacientů homozygotní pro alelu UGT1A1 *28 (n = 112) byl výskyt neutropenie třídy 3-4 58%. U pacientů heterozygotní pro alelu UGT1A1*28 (n = 420) byl výskyt neutropenie třídy 3-4 49%. U pacientů homozygotní pro alelu divokého typu (n = 416) byl výskyt neutropenie třídy 3-4 43% [viz viz Klinická farmakologie ]. In patients homozygous for the UGT1A1 *28 allele the incidence of Grade 3-4 anémie was 21%.
U pacientů heterozygotní pro alelu UGT1A1*28 byla výskyt anémie třídy 3-4 10%. U pacientů homozygotní pro alelu divokého typu byl výskyt anémie třídy 3-4 9%.
Střední čas na první neutropenii včetně febrilní neutropenie byl 9 dní u pacientů homozygotních pro alelu UGT1A1*28 u pacientů heterozygotních pro alelu UGT1A1*28 a 20 dní u pacientů homozygotních pro alelu divokého typu. Střední čas do první anémie byl 21 dní u pacientů homozygotní pro alelu UGT1A1*28 u pacientů heterozygotních pro alelu UGT1A1*28 a 28 dní u pacientů homozygotních pro alelu divokého typu.
Pečlivě sledujte pacienty se známou sníženou aktivitou UGT1A1 pro nežádoucí účinky. Zadržení nebo trvale přerušit Trodelvy na základě klinického posouzení trvání nástupu a závažnosti pozorovaných nežádoucích účinků u pacientů s důkazem akutního včasného nástupu nebo neobvykle závažných nežádoucích účinků, které mohou naznačovat sníženou enzymovou aktivitu UGT1A1 [viz viz aktivita enzymu UGT1A1 [viz viz aktivita enzymu UGT1A1 [viz viz [viz aktivita enzymu UGT1A1 [Viz [viz aktivita enzymu UGT1A1 [viz [viz enzymatická aktivita [ Dávkování a podávání ].
Toxicita embryo-fetální
Na základě svého mechanismu účinku může Trodelvy způsobit teratogenitu a/nebo embryo-fetální letalitu, když je podána těhotné ženě. Trodelvy obsahuje genotoxickou složku SN-38 a cíle rychle dělící buňky [viz Klinická farmakologie a Neklinická toxikologie ]. Admyse pregnant women a females of reproductIVe potential of the potential risk to a fetus. Admyse females of reproductIVe potential to use effectIVe contraception during treatment with Trodelvy a for 6 months after the last dose. Admyse male patients with female partners of reproductIVe potential to use effectIVe contraception during treatment with Trodelvy a for 3 months after the last dose [vidět Použití v konkrétních populacích ].
Informace o poradenství pro pacienta
Doporučujte pacientovi, aby si přečetl značení pacienta schváleného FDA ( Informace o pacientu )
Neutropenie
Poraďte pacientům o riziku neutropenie. Poskytněte pacientům, aby okamžitě kontaktovali svého poskytovatele zdravotní péče, pokud zažívají zimnici nebo jiné známky infekce [viz VAROVÁNÍ AND OPATŘENÍ ].
Průjem
Poraďte pacientům o riziku průjmu. Poskytněte pacientům, aby okamžitě kontaktovali svého poskytovatele zdravotní péče, pokud během léčby poprvé zažijí průjem; černá nebo krvavá stolička; Příznaky dehydratace, jako je závratě nebo slabost; neschopnost brát tekutiny ústy kvůli nevolnosti nebo zvracení; nebo neschopnost dostat průjem pod kontrolou do 24 hodin [viz VAROVÁNÍ AND OPATŘENÍ ].
Přecitlivělost a reakce související s infuzí
Informujte pacienty o riziku závažných infuzních reakcí a anafylaxe. Pokyn pacientům, aby okamžitě kontaktovali svého poskytovatele zdravotní péče, pokud zažívají jazyk na rty nebo otok v krku Potíže s dýcháním závratu závratě zmlácené přísnosti Pruritus Flushing vyrážka nebo horečka, ke které dochází během nebo do 24 hodin po infuzi [Viz viz VAROVÁNÍ AND OPATŘENÍ ].
Nevolnost/Zvracení
Poraďte pacientům o riziku nevolnosti a zvracení. Doporučuje se také premedikace podle zavedených pokynů se dvěma nebo třemi léčivými režimy pro prevenci nevolnosti a zvracení vyvolané chemoterapií (CINV). Jak je klinicky uvedeno, mohou být také použita další antiemetická sedativa a další podpůrná opatření. Všichni pacienti by měli dostávat léky v domácnosti pro prevenci a léčbu zpožděné nevolnosti a zvracení s jasnými pokyny. Poskytněte pacientům, aby okamžitě kontaktovali svého poskytovatele zdravotní péče, pokud zažijí nekontrolovanou nevolnost nebo zvracení [viz VAROVÁNÍ AND OPATŘENÍ ].
Toxicita embryo-fetální
Doporučujte pacientům, aby kontaktovaly svého poskytovatele zdravotní péče, pokud jsou těhotné nebo otěhotní. Informovat pacienty o riziku plodu a potenciální ztrátě těhotenství [viz Použití v konkrétních populacích ].
Antikoncepce
Poraďte pacientům s reprodukčním potenciálem k použití účinné antikoncepce během léčby a po dobu 6 měsíců po poslední dávce Trodelvy [viz Použití v konkrétních populacích ].
Poraďte se s mužskými pacienty s partnery reprodukčního potenciálu k používání účinné antikoncepce během léčby a po dobu 3 měsíců po poslední dávce Trodelvy [viz Použití v konkrétních populacích ].
Laktace
Doporučujte ženám, aby nedošlo k kojení během léčby a po dobu 1 měsíce po poslední dávce Trodelvy [viz Použití v konkrétních populacích ].
Neplodnost
Radí ženám reprodukčního potenciálu, aby Trodelvy mohl narušit plodnost [viz Použití v konkrétních populacích ].
Neklinická toxikologie
Zhodnocení mutageneze karcinogeneze plodnosti
Studie karcinogenity nebyly provedeny u Sacituzumab govitecan-hziy.
SN-38 byl klastogenní v testu mikronukleusových testů in vitro v buňkách vaječníků čínského křečka a nebyl mutagenní v testu bakteriální reverzní mutace (AMES) in vitro.
Studie plodnosti se Sacituzumab govitecan-hziy nebyly provedeny. In a repeat-dose toxicity study in cynomolgus monkeys intravenous administration of sacituzumab govitecan-hziy on Day 1 and Day 4 resulted in endometrial atrophy uterine hemorrhage increased follicular atresia of the ovary and atrophy of vaginal epithelial cells at doses ≥ 60 mg/kg (≥ 6 times the human recommended dose of 10 mg/kg na základě tělesné hmotnosti).
Použití v konkrétních populacích
Těhotenství
Shrnutí rizika
Na základě svého mechanismu účinku může Trodelvy způsobit teratogenitu a/nebo embryo-fetální letalitu, když je podána těhotné ženě. U těhotných žen nejsou k dispozici žádné údaje, které by informovaly o riziku spojeném s drogami. Trodelvy obsahuje genotoxickou složku SN-38 a je toxický pro rychle dělící buňky [viz Klinická farmakologie a Neklinická toxikologie ]. Admyse pregnant women a females of reproductIVe potential of the potential risk to a fetus.
Odhadované riziko na pozadí hlavních vrozených vad a potratu pro uvedenou populaci není známo. V americké obecné populaci je odhadované riziko na pozadí hlavních vrozených vad a potratu u klinicky uznávaných těhotenství 2–4% a 15–20%.
Data
Údaje o zvířatech
Nebyly provezeny žádné reprodukční a vývojové toxikologické studie se Sacituzumab GoviteCan-Hziy.
Laktace
Shrnutí rizika
Neexistují žádné informace týkající se přítomnosti Sacituzumab govitecan-hziy nebo SN-38 v lidském mléce účinky na kojené dítě nebo účinky na produkci mléka. Vzhledem k potenciálu vážných nežádoucích účinků u kojeného dítěte radí ženám, aby se během léčby a po dobu poslední dávky Trodelvy nestaly kojit během léčby a po dobu 1 měsíce.
Ženy a muži reprodukčního potenciálu
Těhotenství Testing
Ověřte stav těhotenství žen reprodukčního potenciálu před zahájením Trodelvy.
Antikoncepce
Ženy
Trodelvy can cause fetal harm when administered to a pregnant woman [vidět Použití v konkrétních populacích ]. Admyse females of reproductIVe potential to use effectIVe contraception during treatment with Trodelvy a for 6 months after the last dose.
Muži
Kvůli potenciálu pro genotoxicitu radí pacientům s ženskými partnery s partnery reprodukčního potenciálu k použití účinné antikoncepce během léčby s Trodelvy a po dobu 3 měsíců po poslední dávce.
Neplodnost
Ženy
Na základě nálezů u zvířat může Trodelvy narušit plodnost u žen s reprodukčním potenciálem [viz Neklinická toxikologie ].
Dětské použití
U pediatrických pacientů nebyla stanovena bezpečnost a účinnost Trodelvy.
Geriatrické použití
Z 366 pacientů s TNBC, kteří byli léčeni trodelvy 19% pacientů, bylo 65 let a 3% bylo 75 let a starších. Pacienti 65 a starší měli zvýšený výskyt neutropenie včetně fatálních výsledků. Nebyly pozorovány žádné další rozdíly v bezpečnosti a účinnosti mezi pacienty ≥ 65 let a mladšími pacienty.
Z 322 pacientů s rakovinou prsu HR /HER2, kteří byli léčeni Trodelvy 26% pacientů, bylo 65 let a starší a 6% bylo 75 let a starších. Nebyly pozorovány žádné celkové rozdíly v účinnosti mezi pacienty ≥ 65 let a mladšími pacienty. U pacientů ve věku 65 let nebo starších (14%) ve srovnání s mladšími pacienty (3%) došlo k vyšší míře přerušení v důsledku nežádoucích účinků ve věku 65 let a starších (14%).
Poškození jater
Při podávání Trodelvy pacientům s mírným poškozením jater není nutné žádné úpravy počáteční dávky [viz viz Klinická farmakologie ].
Nebyla stanovena bezpečnost Trodelvy u pacientů se středním (celkový bilirubin> 1,5 až 3,0 x ULN) nebo těžký (celkový bilirubin> 3,0 x horní hranice normálního [ULN]). Trodelvy nebyl testován u pacientů s AST nebo ALT> 3 Uln bez jaterní metastázy nebo AST nebo ALT> 5 Uln s metastázami jater. Pro počáteční dávku u těchto pacientů nelze učinit žádná doporučení.
Informace o předávkování pro Trodelvy
V klinické studii plánované dávky až 18 mg/kg (přibližně 1,8násobek maximální doporučené dávky 10 mg/kg) byly podány trodelvy. U těchto pacientů byl pozorován vyšší výskyt těžké neutropenie.
Kontraindikace pro Trodelvy
Trodelvy is contraindicated in patients who have experienced a severe hypersensitIVity reaction to Trodelvy [vidět VAROVÁNÍ AND OPATŘENÍ ].
Klinická farmakologie for Trodelvy
Mechanismus působení
SACITUZUMAB GOVITECAN-HZIY je konjugát protilátek s protilátkou TROP-2. Sacituzumab je humanizovaná protilátka, která rozpoznává Trop-2. Malá molekula SN-38 je inhibitor topoisomerázy I, který je kovalentně připojen k protilátce linkerem. Farmakologická data naznačují, že Sacituzumab govitecan-hziy se váže na rakovinné buňky exprimující TROP-2 a je internalizován následným uvolňováním SN-38 hydrolýzou linkeru. SN-38 interaguje s topoisomerázou I a zabraňuje opětovnému ligaci topoisomerázy I-indukovaných zlomů jednotlivých pramenů. Výsledné poškození DNA vede k apoptóze a buněčné smrti. SACITUZUMAB GOVITECAN-HZIY Snížil růst nádoru v modelech xenoštěpu myších trojitých negativních rakoviny prsu.
Farmakodynamika
Vztahy s expozicí-odpovědí na expozice a farmakodynamický časový průběh účinnosti nebyly plně charakterizovány.
Srdeční elektrofyziologie
Maximální průměrná změna oproti základní linii byla 9,7 ms (horní hranice oboustranného 90% intervalu spolehlivosti je 16,8 ms) v doporučené dávce. Mezi zvýšením QTC a koncentracemi SN-38 byl pozorován pozitivní vztah odpovědí na expozici.
Farmakokinetika
Sérová farmakokinetika Sacituzumabu govitecan-hziy a SN-38 byla hodnocena u pacientů s MBC, kteří dostávali Sacituzumab govitecan-hziy jako jediný činidlo v dávce 10 mg/kg. Farmakokinetické parametry Sacituzumab govitecan-hziy a Free SN-38 jsou uvedeny v tabulce 9.
Tabulka 9: Shrnutí průměrných parametrů PK (CV%) Sacituzumab govitecan-hziy a zdarma SN-38**
| Sacituzumab govitecan-hziy (N = 693) | SN-38 zdarma (N = 681) | |
| Cmax [ng/ml] | 239000 (11%) | 98,0 (45%) |
| AUC0-168 [of*H/ML] | 5640000 (22%) | 3696 (56%) |
| *Parametry odhadované na základě analýz populace PK CMAX: Maximální koncentrace séra od 0 do 68 hodin po první dávce AUC0-168: Plocha pod křivkou koncentrace séra po dobu 168 hodin po první dávce |
Rozdělení
Na základě populace farmakokinetická analýza je v ustáleném stavu distribuce distribuce Sacituzumab govetican-hziy 3,6 l.
Odstranění
Střední poločas eliminace (T½) Sacituzumab govitecan-hziy a volný SN-38 u pacientů s metastatickým trojitým negativním karcinomem prsu byl 23,4 a 17,6 hodin. Na základě populační farmakokinetické analýzy je odhadovaná průměrná (%CV) clearance Sacituzumab govitecan-hziy 0,13 l/h (12%).
Metabolismus
Nebyly provedeny žádné studie metabolismu se Sacituzumab GoviteCan-Hziy. SN-38 (skupina Small Molekula Sacituzumabu govitecan-hziy) je metabolizována přes UGT1A1. Metabolit glukuronidu SN-38 (SN-38G) byl detekovatelný v séru pacientů.
Konkrétní populace
Farmakokinetické analýzy u pacientů léčených Trodelvy neidentifikovaly účinek rasy věku (27 až 88 let) (bílá černá nebo asijská) nebo mírná porucha ledvin na střední snížení ledvin (CLCR 30 až 89 ml/min) na farmakokinetiku Sacituzumab Govitecan. Je známo, že eliminace ledvin přispívá minimálně k vylučování SN-38 s malou molekulovou skupinou Sacituzumab govitecan-hziy. Neexistují žádné údaje o farmakokinetice Sacituzumab govitecan-hziy u pacientů se závažným poškozením ledvin (CLCR 15 až 29 ml/min) nebo onemocnění ledvin v konečném stádiu (CLCR <15 mL/min).
Pacienti s poškozením jater
Expozice Sacituzumab govitecan-hziy je podobná u pacientů s mírným jaterním poškozením (celkový bilirubin ≤ uln s Ast> uln nebo bilirubin> 1,0 až ≤ 1,5 ULN s jakýmkoli AST; n = 257) pro pacienty s normální hepatickou funkcí (celkový bilirubin a Ast a Ast U pacientů se středním (celkovým bilirubinem> 1,5 až 3,0 x ULN) nebo závažným (celkový bilirubin> 3,0 x ULN) jaterní zhoršení nejsou známy Sacituzumab govitecan-hziy a bezplatné expozice SN-38. Nebyly provedeny žádné studie interakce léčiva léčiva s Sacituzumabem govitecan-hziy nebo jeho složkami. Inhibitory nebo induktory UGT1A1 mohou zvýšit nebo snížit expozici SN-38 [viz viz Lékové interakce ]. SN-38 je metabolizován přes UGT1A1 [viz Klinická farmakologie ]. Genetic variants of the UGT1A1 gene such as the UGT1A1*28 allele lead to reduced UGT1A1 enzyme actIVity. IndIViduals who are homozygous or heterozygous for the UGT1A1*28 allele are at increased risk for Neutropenie febrile Neutropenie a anémie from Trodelvy compared to indIViduals who are wildtype (*1/*1) [vidět VAROVÁNÍ AND OPATŘENÍ ]. Approximately 20% of the Black or African American population 10% of the White population a 2% of the East Asian population are homozygous for the UGT1A1*28 allele (*28/*28). Approximately 40% of the Black or African American population 50% of the White population a 25% of the East Asian population are heterozygous for the UGT1A1*28 allele (*1/*28). Decreased function alleles other than UGT1A1*28 may be present in certain populations. Detekce tvorby protilátek je vysoce závislá na citlivosti a specificitě testu. Rozdíly v metodách testu vylučují smysluplné srovnání výskytu protilátek proti drogru ve studiích popsaných níže s výskytem protilátek protilátek v jiných studiích, včetně studií Trodelvy. Během mediálního čtyřměsíčního léčebného období napříč klinickými studiemi u pacientů léčených Trodelvy 9 (1,1%) 785 pacientů se vyvinula protilátky proti Sacituzumab Govitecan; 6 z těchto pacientů (0,8% všech pacientů léčených Trodelvy) mělo neutralizující protilátky proti Sacituzumabu Govitecan. Kvůli nízkému výskytu protilátek proti drogám není znám účinek těchto protilátek na farmakokinetiku farmakodynamiky a/nebo účinnost govitekanu Sacituzumabu. Účinnost byla hodnocena v multicentrické otevřené randomizované studii (Ascent; NCT02574455) prováděné u 529 pacientů s neresekovatelným lokálně pokročilým nebo metastatickým trojitým negativním rakovinou prsu (MTNBC), kteří se v průběhu neoadjuvantního nebo adjuvantu (MTNBC) vyskytovali za předpokladu, která byla založena na progresi). Všichni pacienti dostávali předchozí taxanovou léčbu buď v adjuvantní neoadjuvantní nebo pokročilém stádiu, pokud nedošlo k kontraindikaci nebo nesnášenlivosti taxanům během nebo na konci prvního taxanového cyklu. Před zápisem u pacientů se známými nebo podezřelými mozkovými metastázami bylo zapotřebí magnetické rezonance (MRI) pro stanovení mozkových metastáz. Pacienti s mozkovými metastázami se nechali přihlásit až do předdefinovaného maxima 15% pacientů ve výstupní studii. Vyloučeni byli pacienti se známou Gilbertovou chorobou nebo onemocněním pouze pro kosti. Pacienti byli randomizováni (1: 1), aby dostávali trodelvy 10 mg/kg jako intravenózní infuzi ve dnech 1 a 8 z 21denního (n = 267) nebo výběru chemoterapie jednoho činidla (n = 262). Chemoterapie jednoho činidla byla stanovena vyšetřovatelem před randomizací z jedné z následujících možností: eribulin (n = 139) kapecitabin (n = 33) gemcitabin (n = 38) nebo vinorelbin (n = 52). Pacienti byli léčeni až do progrese onemocnění nebo nepřijatelná toxicita. Hlavním výsledkem účinnosti bylo přežití bez progrese (PFS) u pacientů bez mozkových metastáz na začátku (tj. BMNEG), měřeno zaslepeným nezávislým centralizovaným přezkumem hodnoceným pomocí kritérií hodnocení odpovědí u kritérií pevných nádorů (RECIST) v1.1. Mezi další měření účinnosti patřila PFS pro celou populaci (všichni pacienti s mozkovými metastázami a bez mozku) a celkové přežití (OS). Střední věk pacientů v plné populaci (n = 529) byl 54 let (rozmezí: 27 až 82 let); 99,6% byly ženy; 79% bílých 12% bylo černých/afrických Američanů; a 81% pacientů bylo <65 years of age. All patients had an ECOG performance status of 0 (43%) or 1 (57%). Forty-two percent of patients had hepatic metastases 9% were BRCA1/BRCA2 mutational status positIVe a 70% were TNBC at diagnosis. Twelve percent had baseline brain metastases premyously treated a stable (n=61; 32 on Trodelvy arm a 29 on single agent chemotherapy arm). Overall 29% of patients had receIVed prior PD-1/PD-L1 therapy. Thirteen percent of patients in the Trodelvy group in the full population receIVed only 1 prior line of systemic therapy in the metastatic setting. Výsledky účinnosti jsou shrnuty v tabulce 10 a jsou uvedeny na obrázku 1 a na obrázku 2. Výsledky účinnosti pro podskupinu pacientů, kteří dostávali pouze 1 předchozí linii systémové terapie v metastatickém prostředí (kromě recidivy nebo progrese onemocnění do 12 měsíců od neoadjuvantní/adjuvantní systémové terapie) byly v souladu s těmi, kteří v metastatickém nastavení dostali nejméně dvě předchozí linie. Tabulka 10: Výsledky účinnosti z výstupu Obrázek 1: Kaplan-meier graf PFS BICR (všichni randomizovaní pacienti) v Ascen Obrázek 2: Kaplan-Meier graf OS (všichni randomizovaní pacienti) v Ascentu Průzkumná analýza PFS u pacientů s dříve léčenými stabilními mozkovými metastázami ukázala stratifikovanou HR 0,65 (95% CI: 0,35 1,22). Střední PFS v rameni Trodelvy byla 2,8 měsíce (95% CI: 1,5 3,9) a střední PFS s chemoterapií jednoho činidla byla 1,6 měsíce (95% CI: 1,3 2,9). Průzkumná analýza OS ve stejné populaci ukázala stratifikovanou HR 0,87 (95% CI: 0,47 1,63). Střední OS v trodelvy rameni byl 6,8 měsíce (95% CI: 4,7 14,1) a medián OS s chemoterapií jednoho činidla byl 7,4 měsíce (95% CI: 4,7 11,1). Immu-132-01 Účinnost trodelvy byla hodnocena v multicentrické studii s jedním ramenem (NCT01631552), která zařadila 108 pacientů s metastatickým trojitým negativním karcinomem prsu (MTNBC), kteří dostali nejméně dvě předchozí protirakovinové terapie pro metastatické onemocnění. Pacienti s objemnou onemocněním definovaní jako hmotnost> 7 cm nebyli způsobilí. Pacienti s léčenými mozkovými metastázami, které nedostaly steroidy s vysokou dávkou (> 20 mg prednisonu nebo ekvivalentu) po dobu nejméně čtyř týdnů, byli způsobilí. Pacienti se známou Gilbertovou chorobou byli vyloučeni. Pacienti dostávali trodelvy 10 mg/kg intravenózně ve dnech 1 a 8 21denního léčebného cyklu. Pacienti byli léčeni trodelvy až do progrese nebo intolerance onemocnění na terapii. Zobrazování nádoru bylo získáno každých 8 týdnů s potvrzujícími skenováním CT/MRI získaných 4-6 týdnů po počáteční částečné nebo úplné reakci až do progrese vyžadující přerušení léčby. Hlavní měření výsledku účinnosti byla vyšetřovatelé hodnocena celková míra odezvy (ORR) pomocí RECIST 1.1 a trvání odpovědi. Střední věk byl 55 let (rozmezí: 31 až 80 let); 87% pacientů bylo mladších než 65 let. Většina pacientů byla žena (99%) a bílá (76%). Při vstupu do studie měli všichni pacienti výkon výkonného výkonu 0 (29%) nebo 1 (71%). Sedmdesát šest procent mělo viscerální onemocnění 42%, pokud jaterní metastázy 56% mělo metastázy plic/pleura a 2% mělo mozkové metastázy. Dvanáct pacientů (11%) mělo v době počáteční diagnózy onemocnění fáze IV. Střední počet předchozích systémových terapií přijatých v metastatickém prostředí byl 3 (rozmezí: 2 až 10). Mezi předchozí chemoterapie v metastatickém prostředí patřily karboplatina nebo cisplatinový (69%) gemcitabin (55%) paclitaxel nebo docetaxel (53%) kapecitabin (51%) eribulin (45%) doxorubicin (24%) vinorelbin (24%) a ixorubicin (24%). Celkově 98% pacientů obdrželo předchozí taxany a 86% obdrželo předchozí antracykliny buď v (NEO) adjuvantním nebo metastatickém prostředí. Tabulka 11 shrnuje výsledky účinnosti. Tabulka 11: Výsledky účinnosti u pacientů s MTNBC v Immu-132-01 Účinnost trodelvy byla hodnocena v multicentrické otevřené značce randomizované studie (Tropics-02; NCT03901339) prováděných u 543 pacientů s neresekovatelným místně pokročilým nebo metastatickým HR-pozitivním HR2-negativním (IHC 0 IHC 1 nebo IHC 2 /ISH-) Prsící rakovina po postupování v jakémkoli místě v jakémkoli místě v jakémkoli místě v jakémkoli místě v jakémkoli místě v jakémkoli negativním her2-negativním. terapie a taxan; Pacienti dostávali v metastatickém prostředí alespoň dvě předchozí chemoterapie (jeden z nich by mohl být v neoadjuvantním nebo adjuvantním prostředí, pokud došlo k recidivě během 12 měsíců). Pacienti byli randomizováni (1: 1), aby dostávali trodelvy 10 mg/kg jako intravenózní infuzi ve dnech 1 a 8 z 21denního cyklu (n = 272) nebo chemoterapie jednoho činidla (n = 271). Chemoterapie jediného činidla byla stanovena vyšetřovatelem před randomizací z jedné z následujících možností: eribulin (n = 130) vinorelbin (n = 63) gemcitabin (n = 56) nebo kapecitabin (n = 22). Randomizace byla stratifikována následujícími faktory: předchozí chemoterapeutické režimy pro metastatické onemocnění (2 vs. 3-4) viscerální metastázy (ano nebo ne) a endokrinní terapii v metastatickém prostředí po dobu nejméně 6 měsíců (ano nebo ne). Pacienti byli léčeni až do progrese onemocnění nebo nepřijatelná toxicita. Podávání Trodelvy bylo povoleno nad rámec progrese onemocnění definované RECIST, pokud byl pacient klinicky stabilní a vyšetřovatel považoval za odvozování klinického přínosu. Míra primárního výsledku účinnosti byla PFS stanovená BICR na RECIST V1.1. Mezi další měření účinnosti patřila OS ORR BICR a DOR BICR. Střední věk pacientů ve studované populaci byl 56 let (rozmezí: 2786 let) 26% pacientů bylo 65 let nebo více. Většina pacientů byla žena (99%); 67% bylo bílé 4% bylo černých a 3% bylo asijských a 26% bylo neznámé rasy. Pacienti dostávali medián 7 (rozmezí: 3 až 17) předchozích systémových režimů v jakémkoli prostředí a 3 (rozmezí: 0 až 8) předchozích systémových chemoterapeutických režimů v metastatickém prostředí. Přibližně 42% pacientů mělo 2 předchozí chemoterapeutické režimy pro léčbu metastatického onemocnění ve srovnání s 58% pacientů, kteří měli 3 až 4 předchozí režimy chemoterapie a všichni pacienti měli výkon výkonné výkonu 0 (45%) nebo 1 (55%). Devadesát pět procent pacientů mělo viscerální metastázy. Většina pacientů dostávala endokrinní terapii v metastatickém prostředí po dobu ≥ 6 měsíců (86%). Trodelvy demonstrated a statistically significant improvement in PFS a OS versus single agent chemotherapy. Výsledky účinnosti jsou shrnuty v tabulce 12 a obrázku 3 a obrázek 4. Tabulka 12: Výsledky účinnosti z Tropics-02 Obrázek 3: Kaplan-Meier graf PFS od BICR v Tropics-02 Obrázek 4: Kaplan-Meier graf OS v Tropics-02 Studie interakce léčiva
Farmakogenomika
Imunogenita
Klinické studie
Lokálně pokročilý nebo metastatický trojitý negativní rakovina prsu
VÝSTUP
Všichni randomizovaní pacienti Trodelvy
n = 267Chemoterapie jednoho činidla
n = 262 Přežití bez progrese1 na bicr Progrese nebo smrt onemocnění (%) 190 (71%) 171 (65%) Střední PFS v měsících (95% CI) 4.8 1.7 (4.1 5.8) (15 2.5) Poměr rizika2 (95% tam) 0,43 (0,35 0,54) P-hodnota <0.0001 Celkové přežití Úmrtí (%) 179 (67%) 206 (79%) Střední OS v měsících (95% CI) 11.8 6.9 (10,5 13.8) (5,9 7.6) Poměr rizika2 (95% tam) 0,51 (0,41 0,62) P-hodnota <0.0001 1 PFS je definován jako čas od data randomizace k datu první progrese radiologického onemocnění nebo smrti v důsledku jakékoli příčiny, podle toho, co nastane na prvním místě.
2 Stratifikovaný test log-rank upravený pro stratifikační faktory: Počet předchozích chemoterapií Přítomnost známých mozkových metastáz při vstupu studie a oblasti.
CI = interval spolehlivostiTrodelvy
(N = 108) Celková míra odezvy i Orr (95% CI) 33,3% (24,6 43,1) Úplná odpověď 2,8% Částečná reakce 30,6% Doba odezvy i Počet respondentů 36 Střední měsíce (95% CI) 7.7 (4.9 10,8) Rozsah měsíců 1,9 30.4 % s trváním ≥ 6 měsíců 55,6% % s trváním ≥ 12 měsíců 16,7% i Hodnocení vyšetřovatele
CI: Interval spolehlivosti
: Označuje probíhajícíLokálně pokročilý nebo metastatický HR pozitivní HER2-negativní rakovina prsu
Studie Tropics-02
Všichni randomizovaní pacienti Trodelvy
n = 272Chemoterapie jednoho činidla
n = 271 Přežití bez progrese Autor: Bicr1 Střední PFS v měsících 5.5 4.0 (95% tam) (4.2 7.0) (3.1 4.4) Poměr rizika (95% tam) 0,661 (0,529 0,826) P-hodnota2 0.0003 Celkové přežití3 Střední OS v měsících 14.4 11.2 (95% tam) (13.0 15.7) (10.1 12.7) Poměr rizika (95% tam) 0,789 (0,646 0,964) P-hodnota2 0.0200 Míra objektivní odezvy3 Autor: Bicr Míra odezvy % (95 % CI) 21.0 (16,3 26.3) 14.0 (10.1 18.7) Poměr pravděpodobnosti (95% CI) 1,625 (1,034 2,555) P-hodnota 0.0348 Trvání odezvy3 (DOR) BICR Střední dor za měsíce 8.1 5.6 (95% tam) (6,7 9.1) (3.8 7.9) 1 PFS je definován jako čas od data randomizace k datu první progrese radiologického onemocnění nebo smrti v důsledku jakékoli příčiny, podle toho, co nastane na prvním místě.
2 Stratifikovaný test log-rank upravený pro stratifikační faktory: předchozí chemoterapeutické režimy pro metastatické onemocnění (2 vs. 3-4) viscerální metastázy (Y/N) a endokrinní terapie v metastatickém prostředí po dobu nejméně 6 měsíců (ano nebo ne).
BICR = zaslepený nezávislý centrální přehled; CI = interval spolehlivosti
3 Druhá prozatímní analýza OS (provedena, když bylo pozorováno 390 OS událostí)
Informace o pacientech pro Trodelvy
Trodelvy®
(Troh-Dell-Vee)
(Sacituzumab govitecan-hziy) pro injekci pro intravenózní použití
Jaké jsou nejdůležitější informace, které bych měl vědět o Trodelvy?
Trodelvy can cause serious side effects including:
- Nízký počet bílých krvinek (neutropenie). Nízký počet bílých krvinek je běžný u Trodelvy a může být někdy závažný a vést k infekcím, které mohou být život ohrožující nebo způsobit smrt již v prvním cyklu léčby. Váš poskytovatel zdravotní péče by měl zkontrolovat počet krevních buněk během léčby Trodelvy a může poskytnout lék, který pomůže zabránit nízkému počtu krvinek v prvním cyklu léčby, pokud máte zvýšené riziko pro rozvoj nízkého Počet bílých krvinek s horečkou (febrile Neutropenie ). Pokud je váš počet bílých krvinek příliš nízký, váš poskytovatel zdravotní péče bude možná nutný odložit léčbu nebo snížit dávku Trodelvy, poskytne vám lék k léčbě nízkého počtu krvinek nebo v některých případech může trvale zastavit Trodelvy. Váš poskytovatel zdravotní péče vám bude možná muset dát antibiotikum Léky, pokud vyvinete horečku, zatímco počet bílých krvinek je nízký.
Okamžitě zavolejte svého poskytovatele zdravotní péče, pokud během léčby vyvinete některý z následujících příznaků infekce:
- horečka
- zimnice
- kašel
- dušnost
- pálení nebo bolest, když můžete močit
- Těžká průjem. Průjem is common with Trodelvy a can also be severe. Severe průjem can lead to loss of too much body fluid (dehydration) a kidney problems. Your healthcare promyder should monitor you for průjem a gIVe you medicine as needed to help control your průjem. If you lose too much body fluid your healthcare promyder may need to gIVe you fluids a electrolytes to replace body salts. If you develop průjem during treatment with Trodelvy your healthcare promyder should check to see if průjem may be caused by an infection. Your healthcare promyder may decrease your dose delay treatment or permanently stop Trodelvy if your průjem is severe a cannot be controlled with anti-průjeml medicines.
Okamžitě zavolejte svému poskytovateli zdravotní péče:
- poprvé, kdy dostanete průjem během léčby Trodelvy
- Pokud máte černou nebo krvavou stolici
- Máte -li příznaky ztráty příliš velkého množství tělesné tekutiny a tělesných solí, jako je závratě nebo slabost
- Pokud nemůžete vzít tekutiny ústy kvůli nevolnosti nebo zvracení
- Pokud nejste schopni dostat svůj průjem pod kontrolu do 24 hodin
Co je Trodelvy?
vedlejší účinky drogového lipitoru
Trodelvy is a prescription medicine used to treat adults with:
- typ rakoviny prsu zvaný trojitý negativní rakovina prsu (TNBC), který je estrogen a progesterone hormone receptor (HR)-negatIVe a human epidermal growth factor receptor 2 (HER2)-negatIVe. Trodelvy may be used:
- Když se vaše rakovina prsu rozšířila do jiných částí těla (metastatického) nebo jej nelze odstranit operací a
- Pokud jste dříve obdrželi dvě nebo více předchozí léčby, včetně alespoň jedné léčby metastatického onemocnění.
- Typ rakoviny prsu, který je HR pozitivní a HER2-negativní. Může být použit trodelvy:
- Když se vaše rakovina prsu rozšířila do jiných částí těla nebo ji nelze odstranit chirurgickým zákrokem a
- Pokud jste dříve podstoupili endokrinní terapii a alespoň dvě další léčba metastatického onemocnění.
Není známo, zda je Trodelvy bezpečný a efektivní u lidí s mírnými nebo těžkými problémy s jatery.
Není známo, zda je Trodelvy u dětí bezpečný a efektivní.
Nedostávejte Trodelvy, pokud jste měli závažnou alergickou reakci na Trodelvy. Pokud si nejste jisti, zeptejte se svého poskytovatele zdravotní péče.
Než obdržíte Trodelvy, řekněte svému poskytovateli zdravotní péče o všech vašich zdravotních stavech, včetně, pokud jste:
- Bylo řečeno, že máte gen pro uridin diffikufát-glukuronosyl defferáza 1A1 (UGT1A1)*28. Lidé, kteří nesou tento gen, mají zvýšené riziko, že získáte vedlejší účinky s trodelvy, zejména s nízkým bílým krvištěm počítá horečku, zatímco počet bílých krvinek je nízký a nízký počet červených krvinek. Vidět Jaké jsou nejdůležitější informace, které bych měl vědět o Trodelvy?
- mít problémy s jatery.
- jsou těhotné nebo plánují otěhotnět. Trodelvy může poškodit vaše nenarozené dítě. Váš poskytovatel zdravotní péče by měl zkontrolovat, zda jste těhotná, než začnete dostávat Trodelvy.
- Ženy who can become pregnant should use effectIVe birth control during treatment a for 6 months after your last dose of Trodelvy. Talk to your healthcare promyder about birth control choices that may be right for you during this time. Tell your healthcare promyder right away if you become pregnant during treatment with Trodelvy.
- Muži with a female partner who can become pregnant should use effectIVe birth control during treatment a for 3 months after your last dose of Trodelvy.
- jsou kojení nebo plánují kojení. Není známo, zda Trodelvy prochází do vašeho mateřského mléka a může poškodit vaše dítě. Během léčby a 1 měsíc po poslední dávce Trodelvy ne kojíte.
Řekněte svému poskytovateli zdravotní péče o všech lécích, které užíváte včetně předpisu a volně prodejných léků vitamínů a bylinných doplňků. Některé léky mohou ovlivnit způsob, jakým Trodelvy funguje.
Jak dostanu Trodelvy?
- Váš poskytovatel zdravotní péče vám dává Trodelvy do vaší žíly přes intravenózní (IV) linii.
- Trodelvy is gIVen 1 time each week on Day 1 a on Day 8 of a 21-day treatment cycle.
- Během 3 hodin obdržíte první dávku Trodelvy. Pokud tolerujete první dávku, budoucí budoucí dávky mohou být podávány po dobu 1 až 2 hodin.
- Před každou dávkou Trodelvy obdržíte léky, které vám pomohou zabránit reakcím souvisejícím s infuzí a nevolností a zvracení.
- Budete monitorovány na vedlejší účinky během a po dobu nejméně 30 minut po obdržení každé infuze Trodelvy.
- Váš poskytovatel zdravotní péče může zpomalit nebo dočasně zastavit infuzi Trodelvy, pokud máte reakci související s infuzí nebo trvale zastavit Trodelvy, pokud máte život ohrožující reakci na infuzi.
- Váš poskytovatel zdravotní péče rozhodne, jak dlouho budete i nadále dostávat Trodelvy.
Jaké jsou možné vedlejší účinky Trodelvy?
Trodelvy can cause serious side effects including:
- Vidět Jaké jsou nejdůležitější informace, které bych měl vědět o Trodelvy?
- Alergické a infuzní reakce. Trodelvy can cause serious or life-threatening allergic a infusion-related reactions. Tell your healthcare promyder or nurse right away if you get any of the folnízkýing symptoms of an allergic or infusion-related reaction during your infusion of Trodelvy or within 24 hours after you receIVe a dose of  Trodelvy:
- Otok vašeho obličeje rty jazyk nebo krk
- kopřivka
- svědění nebo spláchnutí pokožky pokožky
- horečka
- potíže s dýcháním or wheezing
- Lightheadedness závrať feeling faint or pass out
- zimnice or shaking zimnice (rigors)
- Nevolnost a zvracení. Nevolnost a zvracení are common with Trodelvy a can sometimes be severe. Before each dose of Trodelvy you will receIVe medicines to help prevent nevolnost a zvracení. You should be gIVen medicines to take home with you along with instructions about how to take them to help prevent a treat any nevolnost a zvracení after you receIVe Trodelvy. Call your healthcare promyder right away if you have nevolnost or zvracení that is not controlled with the medicines prescribed for you. Your healthcare promyder may decide to decrease your dose delay treatment or permanently stop Trodelvy if your nevolnost a zvracení is severe a cannot be controlled with anti-nevolnost medicines.
Mezi nejčastější vedlejší účinky Trodelvy patří:
- snížené bílé krvinky (leukocyt a lymfocyty) a počet červených krvinek
- cítit se unavený nebo slabý
- Vypadávání vlasů
- zácpa
- zvýšená hladina cukru v krvi
- snížené hladiny proteinu (albumin) v
- snížená chuť k jídlu
- Změny v testu funkce ledvin
- Zvýšené hladiny enzymu zvaného alkalická fosfatáza v krvi (test na problémy s játry nebo kostí)
- snížené hladiny draslíku a sodíku hořčíku v krvi
Trodelvy may cause fertility problems in females which could affect your ability to have a baby. Talk to your healthcare promyder if fertility is a concern for you.
Nejedná se o všechny možné vedlejší účinky Trodelvy.
Zavolejte svého lékaře, kde najdete lékařskou radu ohledně vedlejších účinků. Můžete nahlásit vedlejší účinky FDA na 1-800-FDA-1088.
Obecné informace o bezpečném a efektivním používání Trodelvy.
Léky jsou někdy předepsány pro jiné účely než ty, které jsou uvedeny v letáku informací o pacientech. Můžete požádat svého lékárníka nebo poskytovatele zdravotní péče o informace o Trodelvy, která je psána pro zdravotnické pracovníky.
Jaké jsou ingredience v Trodelvy?
Aktivní složka: Sacituzumab govitecan-hziy
Neaktivní ingredience: 2- (N-morfolino) polysorbát kyseliny ethanové sulfonové (MES) 80 a trehalóza
Informace o pacientovi byly schváleny americkou správou potravin a léčiv.